

Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS)
- PMID: 11818550
- PMCID: PMC122237
- DOI: 10.1073/pnas.032539299
Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS)
Abstract
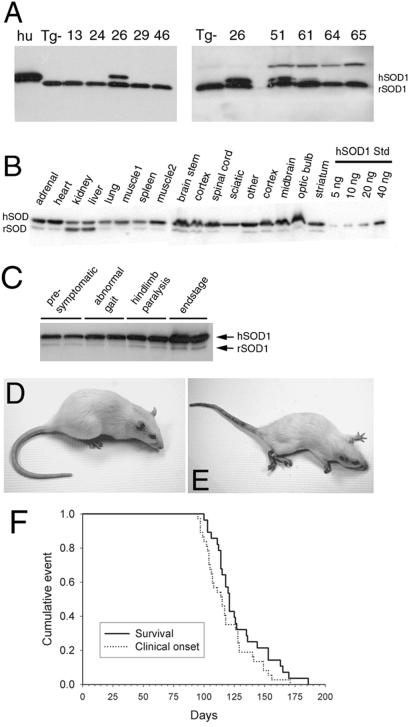
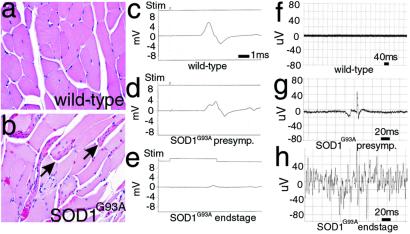
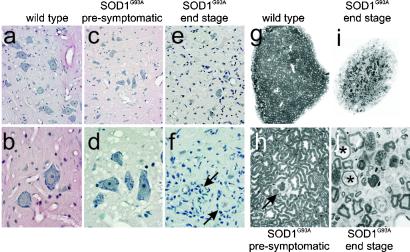
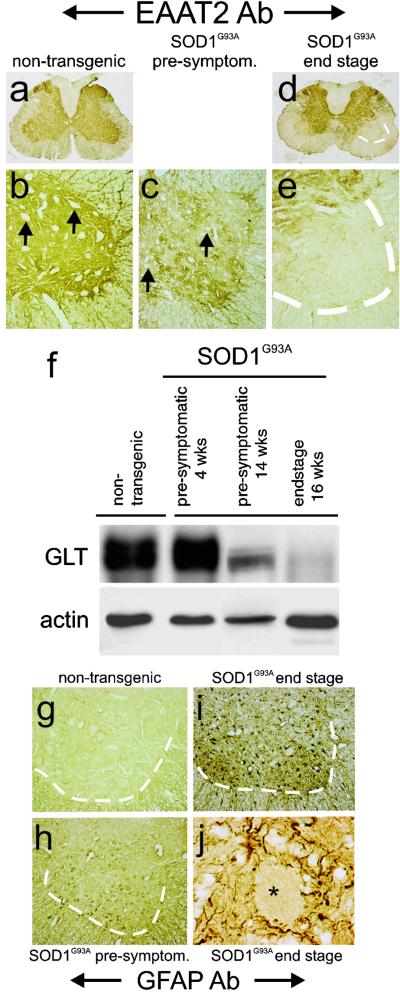
Transgenic overexpression of Cu(+2)/Zn(+2) superoxide dismutase 1 (SOD1) harboring an amyotrophic lateral sclerosis (ALS)-linked familial genetic mutation (SOD1(G93A)) in a Sprague-Dawley rat results in ALS-like motor neuron disease. Motor neuron disease in these rats depended on high levels of mutant SOD1 expression, increasing from 8-fold over endogenous SOD1 in the spinal cord of young presymptomatic rats to 16-fold in end-stage animals. Disease onset in these rats was early, approximately 115 days, and disease progression was very rapid thereafter with affected rats reaching end stage on average within 11 days. Pathological abnormalities included vacuoles initially in the lumbar spinal cord and subsequently in more cervical areas, along with inclusion bodies that stained for SOD1, Hsp70, neurofilaments, and ubiquitin. Vacuolization and gliosis were evident before clinical onset of disease and before motor neuron death in the spinal cord and brainstem. Focal loss of the EAAT2 glutamate transporter in the ventral horn of the spinal cord coincided with gliosis, but appeared before motor neuron/axon degeneration. At end-stage disease, gliosis increased and EAAT2 loss in the ventral horn exceeded 90%, suggesting a role for this protein in the events leading to cell death in ALS. These transgenic rats provide a valuable resource to pursue experimentation and therapeutic development, currently difficult or impossible to perform with existing ALS transgenic mice.
Figures

Similar articles
-
Impaired spinal cord glutamate transport capacity and reduced sensitivity to riluzole in a transgenic superoxide dismutase mutant rat model of amyotrophic lateral sclerosis.J Neurosci. 2003 Mar 1;23(5):1688-96. doi: 10.1523/JNEUROSCI.23-05-01688.2003. J Neurosci. 2003. PMID: 12629173 Free PMC article.
-
Increased expression of the glial glutamate transporter EAAT2 modulates excitotoxicity and delays the onset but not the outcome of ALS in mice.Hum Mol Genet. 2003 Oct 1;12(19):2519-32. doi: 10.1093/hmg/ddg267. Epub 2003 Aug 5. Hum Mol Genet. 2003. PMID: 12915461
-
GLT1 overexpression in SOD1(G93A) mouse cervical spinal cord does not preserve diaphragm function or extend disease.Neurobiol Dis. 2015 Jun;78:12-23. doi: 10.1016/j.nbd.2015.03.010. Epub 2015 Mar 25. Neurobiol Dis. 2015. PMID: 25818008
-
Role of complement in motor neuron disease: animal models and therapeutic potential of complement inhibitors.Adv Exp Med Biol. 2008;632:143-58. Adv Exp Med Biol. 2008. PMID: 19025120 Review.
-
[Amyotrophic lateral sclerosis with the SOD1 mutations].Rinsho Shinkeigaku. 2008 Nov;48(11):966-9. doi: 10.5692/clinicalneurol.48.966. Rinsho Shinkeigaku. 2008. PMID: 19198133 Review. Japanese.
Cited by
-
All-in-one wearable drug efficacy assessment systems for bulbar muscle function using amyotrophic lateral sclerosis animal models.Nat Commun. 2024 Aug 9;15(1):6803. doi: 10.1038/s41467-024-51300-1. Nat Commun. 2024. PMID: 39122743 Free PMC article.
-
From animal models to human disease: a genetic approach for personalized medicine in ALS.Acta Neuropathol Commun. 2016 Jul 11;4(1):70. doi: 10.1186/s40478-016-0340-5. Acta Neuropathol Commun. 2016. PMID: 27400686 Free PMC article. Review.
-
Synergistic effects of GDNF and VEGF on lifespan and disease progression in a familial ALS rat model.Mol Ther. 2013 Aug;21(8):1602-10. doi: 10.1038/mt.2013.108. Epub 2013 May 28. Mol Ther. 2013. PMID: 23712039 Free PMC article.
-
Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis.Proc Natl Acad Sci U S A. 2006 Oct 24;103(43):16021-6. doi: 10.1073/pnas.0607423103. Epub 2006 Oct 16. Proc Natl Acad Sci U S A. 2006. PMID: 17043238 Free PMC article.
-
Neuroprotective effects of violacein in a model of inherited amyotrophic lateral sclerosis.Sci Rep. 2022 Mar 15;12(1):4439. doi: 10.1038/s41598-022-06470-7. Sci Rep. 2022. PMID: 35292673 Free PMC article.
References
-
- Delisile M B, Carpenter S. J Neurol Sci. 1984;63:241–250. - PubMed
-
- Banker B Q. In: Myology. Engel A G, Banker B Q, editors. New York: McGraw–Hill; 1986. pp. 2031–2066.
-
- Horton W A, Eldredge R, Brody J A. Neurology. 1976;26:460–665. - PubMed
-
- Rosen D R, Siddique T, Patterson D, Figlewicz D A, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan J P, Deng H X, et al. Nature (London) 1993;362:59–62. - PubMed
-
- Deng H X, Hentati A, Tainer J A, Iqbal Z, Cayabyab A, Hung W Y, Getzoff E D, Hu P, Herzfeldt B, Roos R P, et al. Science. 1993;261:1047–1051. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
