

x ray crystallography
- PMID: 10884915
- PMCID: PMC1186895
- DOI: 10.1136/mp.53.1.8
x ray crystallography
Abstract
x Ray crystallography is currently the most favoured technique for structure determination of proteins and biological macromolecules. Increasingly, those interested in all branches of the biological sciences require structural information to shed light on previously unanswered questions. Furthermore, the availability of a protein structure can provide a more detailed focus for future research. The extension of the technique to systems such as viruses, immune complexes, and protein-nucleic acid complexes serves only to widen the appeal of crystallography. Structure based drug design, site directed mutagenesis, elucidation of enzyme mechanisms, and specificity of protein-ligand interactions are just a few of the areas in which x ray crystallography has provided clarification.
Figures

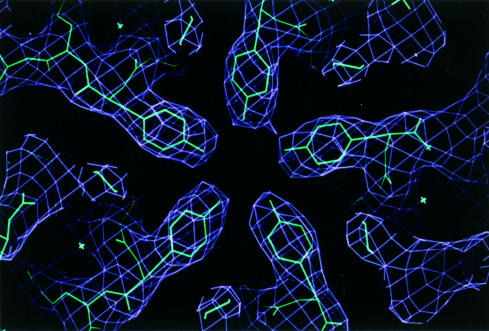
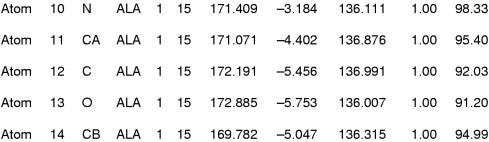
Similar articles
-
2dx--user-friendly image processing for 2D crystals.J Struct Biol. 2007 Jan;157(1):64-72. doi: 10.1016/j.jsb.2006.07.020. Epub 2006 Sep 1. J Struct Biol. 2007. PMID: 17055742
-
X-ray crystallography as a tool for mechanism-of-action studies and drug discovery.Curr Pharm Biotechnol. 2013;14(5):537-50. doi: 10.2174/138920101405131111104824. Curr Pharm Biotechnol. 2013. PMID: 22429136 Review.
-
A general method for the unbiased improvement of solution NMR structures by the use of related X-ray data, the AUREMOL-ISIC algorithm.BMC Struct Biol. 2006 Jun 26;6:14. doi: 10.1186/1472-6807-6-14. BMC Struct Biol. 2006. PMID: 16800891 Free PMC article.
-
Determination of soluble and membrane protein structures by X-ray crystallography.Methods Mol Biol. 2013;955:475-93. doi: 10.1007/978-1-62703-176-9_25. Methods Mol Biol. 2013. PMID: 23132076
-
X-ray crystallography over the past decade for novel drug discovery - where are we heading next?Expert Opin Drug Discov. 2015;10(9):975-89. doi: 10.1517/17460441.2015.1061991. Epub 2015 Jul 15. Expert Opin Drug Discov. 2015. PMID: 26177814 Free PMC article. Review.
Cited by
-
Exploring the World of Membrane Proteins: Techniques and Methods for Understanding Structure, Function, and Dynamics.Molecules. 2023 Oct 19;28(20):7176. doi: 10.3390/molecules28207176. Molecules. 2023. PMID: 37894653 Free PMC article. Review.
-
Deep-learning based 3D birefringence image generation using 2D multi-view holographic images.Sci Rep. 2024 Apr 30;14(1):9879. doi: 10.1038/s41598-024-60023-8. Sci Rep. 2024. PMID: 38684698 Free PMC article.
-
Identifying the minimal sets of distance restraints for FRET-assisted protein structural modeling.Protein Sci. 2024 Dec;33(12):e5219. doi: 10.1002/pro.5219. Protein Sci. 2024. PMID: 39548730
-
Coevolution and smFRET Enhances Conformation Sampling and FRET Experimental Design in Tandem PDZ1-2 Proteins.J Phys Chem B. 2023 Feb 2;127(4):884-898. doi: 10.1021/acs.jpcb.2c06720. Epub 2023 Jan 24. J Phys Chem B. 2023. PMID: 36693159 Free PMC article.
-
Structural Elucidation of Ubiquitin via Gas-Phase Ion/Ion Cross-Linking Reactions Using Sodium-Cationized Reagents Coupled with Infrared Multiphoton Dissociation.Anal Chem. 2024 May 28;96(21):8518-8527. doi: 10.1021/acs.analchem.4c00442. Epub 2024 May 6. Anal Chem. 2024. PMID: 38711366
References
-
- Blundell TL, Johnson LN. Protein crystallography. London: Academic Press, 1976.
-
- Carter CW, Sweet RM, eds. Macromolecular crystallography, part A. Methods Enzymol 1997;276.
-
- Carter CW, Sweet RM, eds. Macromolecular Crystallography, part B. Methods Enzymol 1997;277.
-
- Luft JR, Arakali SV, Kirisits J, et al. A macromolecular crystallization procedure employing diffusion cells of varying depths as reservoirs to taylor the time course of equilibration in hanging drop and sitting drop vapour diffusion and microdialysis experiments. Journal of Applied Crystallography 1994;27:443–53.
-
- Wilson LJ, Bray TL, Suddath FL. Crystallization of proteins by dynamic control of evaporation. Journal of Crystal Growth 1991;110:142–7.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources