

Progressive region-specific de novo methylation of the p16 CpG island in primary human mammary epithelial cell strains during escape from M(0) growth arrest
- PMID: 10409753
- PMCID: PMC84416
- DOI: 10.1128/MCB.19.8.5642
Progressive region-specific de novo methylation of the p16 CpG island in primary human mammary epithelial cell strains during escape from M(0) growth arrest
Abstract
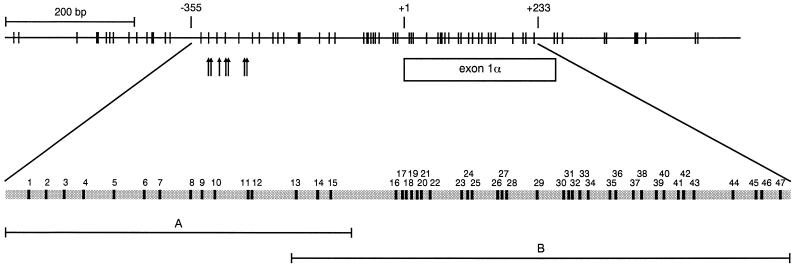
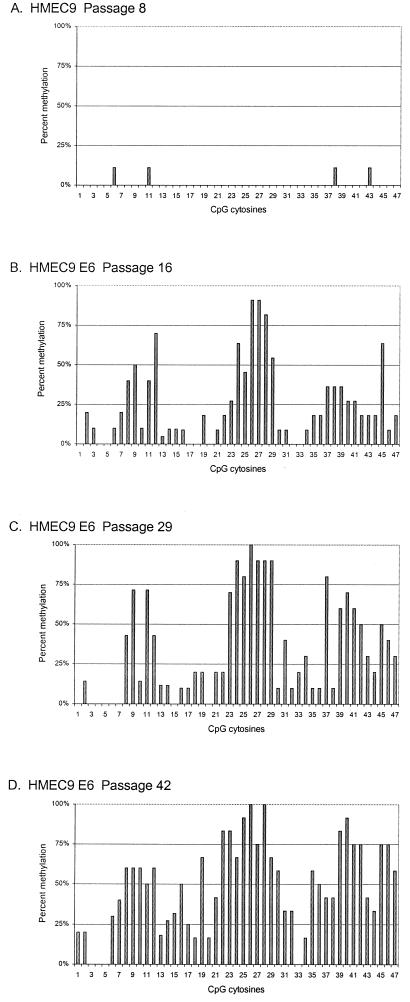
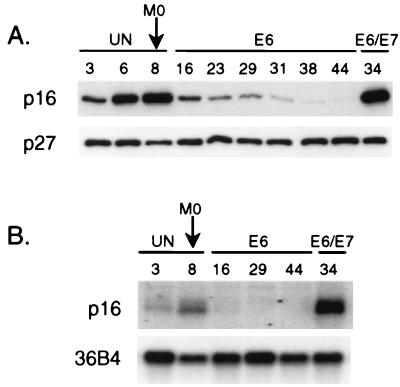
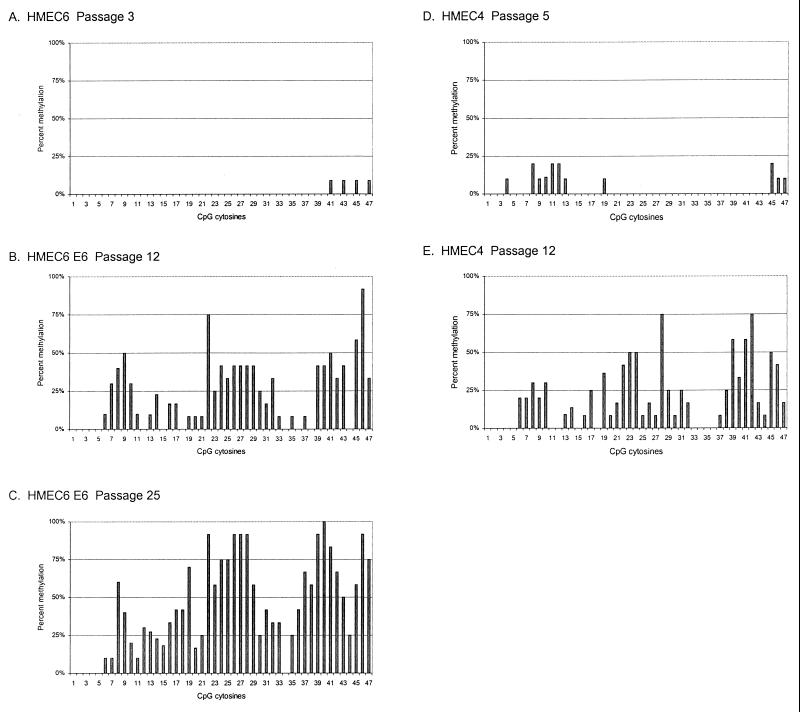
CpG island methylation plays an important role in normal cellular processes, such as genomic imprinting and X-chromosome inactivation, as well as in abnormal processes, such as neoplasia. However, the dynamics of de novo CpG island methylation, during which a CpG island is converted from an unmethylated, active state to a densely methylated, inactive state, are largely unknown. It is unclear whether the development of de novo CpG island methylation is a progressive process, in which a subset of CpG sites are initially methylated with a subsequent increase in methylation density, or a single event, in which the initial methylation event encompasses the entire CpG island. The tumor suppressor gene p16/CDKN2a/INK4a (p16) is inactivated by CpG island methylation during neoplastic progression in a variety of human cancers. We investigated the development of methylation in the p16 CpG island in primary human mammary epithelial cell strains during escape from mortality stage 0 (M(0)) growth arrest. The methylation status of 47 CpG sites in the p16 CpG island on individual DNA molecules was determined by sequencing PCR clones of bisulfite-treated genomic DNA. The p16 CpG island was initially methylated at a subset of sites in three discrete regions in association with p16 transcriptional repression and escape from M(0) growth arrest. With continued passage, methylation gradually increased in density and methylation expanded to sites in adjacent regions. Thus, de novo methylation in the p16 CpG island is a progressive process that is neither site specific nor completely random but instead is region specific. Our results suggest that early detection of methylation in the CpG island of the p16 gene will require methylation analysis of the three regions and that the identification of region-specific methylation patterns in other genes may be essential for an accurate assessment of methylation-mediated transcriptional silencing.
Figures

Similar articles
-
Aberrant de novo methylation of the p16INK4A CpG island is initiated post gene silencing in association with chromatin remodelling and mimics nucleosome positioning.Hum Mol Genet. 2009 Aug 15;18(16):3098-109. doi: 10.1093/hmg/ddp251. Epub 2009 May 28. Hum Mol Genet. 2009. PMID: 19477956
-
Mapping patterns of CpG island methylation in normal and neoplastic cells implicates both upstream and downstream regions in de novo methylation.J Biol Chem. 1997 Aug 29;272(35):22322-9. doi: 10.1074/jbc.272.35.22322. J Biol Chem. 1997. PMID: 9268383
-
Nucleosomes correlate with in vivo progression pattern of de novo methylation of p16 CpG islands in human gastric carcinogenesis.PLoS One. 2012;7(4):e35928. doi: 10.1371/journal.pone.0035928. Epub 2012 Apr 25. PLoS One. 2012. PMID: 22558275 Free PMC article.
-
Decreased fidelity in replicating DNA methylation patterns in cancer cells leads to dense methylation of a CpG island.Curr Top Microbiol Immunol. 2006;310:199-210. doi: 10.1007/3-540-31181-5_10. Curr Top Microbiol Immunol. 2006. PMID: 16909912 Review.
-
DNA methylation in poultry: a review.J Anim Sci Biotechnol. 2023 Nov 5;14(1):138. doi: 10.1186/s40104-023-00939-9. J Anim Sci Biotechnol. 2023. PMID: 37925454 Free PMC article. Review.
Cited by
-
NSAIDs modulate CDKN2A, TP53, and DNA content risk for progression to esophageal adenocarcinoma.PLoS Med. 2007 Feb;4(2):e67. doi: 10.1371/journal.pmed.0040067. PLoS Med. 2007. PMID: 17326708 Free PMC article.
-
Bmi1, stem cells, and senescence regulation.J Clin Invest. 2004 Jan;113(2):175-9. doi: 10.1172/JCI20800. J Clin Invest. 2004. PMID: 14722607 Free PMC article. Review.
-
Overexpression of RhoA induces preneoplastic transformation of primary mammary epithelial cells.Cancer Res. 2009 Jan 15;69(2):483-91. doi: 10.1158/0008-5472.CAN-08-2907. Cancer Res. 2009. PMID: 19147561 Free PMC article.
-
Mapping of the methylation pattern of the hMSH2 promoter in colon cancer, using bisulfite genomic sequencing.J Carcinog. 2006 Aug 15;5:22. doi: 10.1186/1477-3163-5-22. J Carcinog. 2006. PMID: 16911791 Free PMC article.
-
Cloning of the 5' upstream region of the rat p16 gene and its role in silencing.Jpn J Cancer Res. 2002 Oct;93(10):1100-6. doi: 10.1111/j.1349-7006.2002.tb01211.x. Jpn J Cancer Res. 2002. PMID: 12417039 Free PMC article.
References
-
- An H X, Niederacher D, Picard F, van Roeyen C, Bender H G, Beckmann M W. Frequent allele loss on 9p21-22 defines a smallest common region in the vicinity of the CDKN2 gene in sporadic breast cancer. Genes Chromosomes Cancer. 1996;17:14–20. - PubMed
-
- Baylin S B, Herman J G, Graff J R, Vertino P M, Issa J P. Alterations in DNA methylation: a fundamental aspect of neoplasia. Adv Cancer Res. 1998;72:141–196. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous