

Abstract
Numerous proteins, including cytokines and chemokines, enzymes and enzyme inhibitors, extracellular matrix proteins, and membrane receptors, bind heparin. Although they are traditionally classified as heparin-binding proteins, under normal physiological conditions these proteins actually interact with the heparan sulfate chains of one or more membrane or extracellular proteoglycans. Thus, they are more appropriately classified as heparan sulfate–binding proteins (HSBPs). This review provides an overview of the various modes of interaction between heparan sulfate and HSBPs, emphasizing biochemical and structural insights that improve our understanding of the many biological functions of heparan sulfate.
Keywords: heparin-binding protein, glycosaminoglycan, proteoglycan, glycan–protein interaction, heparan sulfate–binding domain, oligomerization
INTRODUCTION
Heparan sulfate (HS) is a linear sulfated glycosaminoglycan (GAG) expressed by virtually all animal cells. It is an ancient molecule that is present in Cnidaria (e.g., Hydra) and all metazoans analyzed to date, with the exception of Porifera (1–3). Amazingly, it has undergone very limited structural variation over ~500 million years of evolution. In contrast, hundreds of proteins have evolved the capacity to interact with HS, often with great specificity. This network of HS-binding proteins (HSBPs), termed the heparan sulfate interactome (4), includes proteins involved in cell attachment, migration, invasion and differentiation, morphogenesis, organogenesis, blood coagulation, lipid metabolism, inflammation, and responses to injury (5). All of these processes involve physical docking of relevant HSBPs to carbohydrate sequences within the HS chains. The absence of HS is not compatible with life (6, 7), a finding that reflects the capacity of HS to bind and modulate the activity of the HSBPs.
Like other macromolecules, HS can be divided into subunits, which are operationally defined as disaccharides on the basis of the ability of bacterial enzymes or nitrous acid to cleave the chain into its component units. The basic building block consists of β1–4-linked d-glucuronic acid (GlcA) and α1–4-linked N-acetyl-d-glucosamine (GlcNAc) (Figure 1). The copolymer assembles while covalently attached to a limited number of proteins, which are referred to as proteoglycan core proteins. Only 17 proteoglycans are known to contain HS (see the sidebar), although other proteoglycans are undoubtedly expressed in a tissue-specific or developmentally regulated manner or in other organisms. During the assembly process, the chains undergo a series of processing reactions in which subsets of N-acetylglucosamine residues become N-deacetylated and N-sulfated; adjacent glucuronic acids undergo epimerization at C5 to l-iduronic acid (IdoA); and ester-linked sulfate groups are installed at C2 of the uronic acids, at C6 of the glucosamine residues, and more rarely at C3 of the glucosamine residues. These modification reactions do not go to completion, giving rise to sections of the chains with variably sulfated domains (so-called NS domains), which are interspersed by NAc domains lacking all or most of these modifications, and mixed NAc/NS domains, which are located in the transition zone between NS and NAc domains (8, 9). Although all animal cells make HS, the size, composition, and distribution of the NS domains vary significantly. Unlike DNA, RNA, and protein assembly, the assembly of HS is not template driven. Thus, the organization and specificity of the biosynthetic enzymes, availability of precursors, and flux through the Golgi apparatus are thought to determine chain length, degree of sulfation, and epimerization, as well as the size and spacing of the sulfated domains. The assembly and degradation of HS have been reviewed elsewhere and are not considered further herein (10–13). What dictates the size and composition of HS in different cells at different times in development remains one of the great enigmas in modern cell biology.
Figure 1.
Structure of heparan sulfate. (a) Chemical structure of a heparin-derived decasaccharide. Carbon numbers of the modification sites are indicated in red. (b) Stereo view of a heparin-derived decasaccharide in space-filling representation, based on Protein Data Bank identifier 1E0O, with carbon in gray, oxygen in red, nitrogen in blue, and sulfate in yellow. (c) Stereo view of the same decasaccharide in stick representation. Note the helical nature of the chain and the alternating clusters of three sulfates groups on each side of the sugar backbone. Abbreviations: GlcNS, N-sulfoglucosamine; IdoA, L-iduronic acid.
Importantly, the terms heparan and heparin are often used interchangeably, which is incorrect. HS occurs naturally in all cells and varies enormously in terms of degree of sulfation and chain length, which depend on its biological origin. The chains typically consist of 50 to 250 disaccharide units (20–100 kDa). In contrast, heparin is a degradation product derived from HS isolated from porcine entrails or equine lung and typically consists of chains ranging from 12 to 14 kDa. Heparin contains larger NS domains and the extent of modification is greater, giving rise to large sections of the chains containing fully sulfated (trisulfated) disaccharides and IdoA (Table 1 summarizes the differences between heparin and HS). The manufacturing process for producing heparin enriches for chains with these characteristics and for the rare 3-O-sulfate groups that drive the interaction of heparin with antithrombin (AT), as explained in greater detail below (in the section titled Specificity of Heparan Sulfate–Protein Interactions). Heparin is the largest biopharmaceutical in production worldwide because of its potent anticoagulant activity. It is also extremely useful for identifying so-called heparin-binding proteins, but the high negative charge of the chains also endows heparin with potent nonspecific cation-exchange properties. Thus, the fact that a protein binds to heparin does not necessarily mean that it also binds to HS.
Table 1.
Differences between heparan sulfate and heparin
| Characteristics | Heparan sulfate | Heparin |
|---|---|---|
| Site of synthesis | Virtually all cells | Connective tissue-type mast cells |
| Core protein | Many (~17) | Serglycin |
| Cell membrane attached? | Yes | No |
| Size | 20–100 kDa | 7–20 kDa |
| Alternate NS/NAc domains | Yes | Minimal |
| Average sulfate/disaccharide | 0.6–1.8 | 1.8–2.6 |
| IdoA | 20–50% | ≥80% |
| N-Sulfate | 30–60% | ≥80% |
| 2-O-Sulfate | 10–40% | ≥80% |
| 6-O-Sulfate | 10–40% | ≥80% |
| Binding to antithrombin | 0–0.3% | ~30% |
| Commercial availability | Milligrams | Kilograms |
Abbreviation: IdoA, l-iduronic acid.
The purpose of this review is to provide an overview of HSBPs and the various ways in which these proteins interact with HS or heparin, drawing on specific examples that illustrate underlying biochemical and structural principles. Many bacteria and viruses also express HSBPs and are discussed elsewhere (14, 15). For brevity, we discuss mostly eukaryotic HSBPs for which the HS-binding sites have been documented by mutagenesis or by cocrystallization studies. We hope that this information will demystify HS–protein interactions and provide insights from a structural perspective into the varied biological functions of HS.
DEFINITION OF A HEPARAN SULFATE–BINDING PROTEIN
As their name suggests, HSBPs are proteins that show appreciable binding to HS at a physiologically relevant ionic strength and pH. As a general rule, the following criteria should be satisfied for a given protein to be referred to as an HSBP. First, the protein should bind to heparin (usually in the form of heparin/Sepharose® beads) and remain bound following washing with a buffer containing isotonic saline. This criterion is a convenient first test because heparin serves as a surrogate for HS and is commercially available in large quantities and as a chromatography resin. Second, the protein should bind to HS in isotonic saline. Third, because HS generally performs its biological functions at the cell surface and in the extracellular matrix, the protein should be present in a relevant biological context. Many proteins in the cytoplasm or the nucleus (e.g., histones, polymerases, transcription factors), although capable of binding to heparin or HS, may not be genuine HSBPs. Some evidence suggests the existence of GAGs in the nucleus, which suggests that the interaction may have biological consequences. Whether HS in this location docks with specific proteins is unclear (16–19). Currently, at least 300 secreted and membrane-associated human proteins are known to bind heparin, and the vast majority of these proteins are expected to be HSBPs (4). This number probably underestimates the total number of HSBPs, given that new members are identified and characterized on a regular basis.
Heparan Sulfate–Binding Proteins Are Structurally Unrelated but Evolutionarily Conserved
HSBPs possess many different structural domains that can form binding sites for HS. Thus, most of the HSBPs appear to be unrelated to each other, suggesting that the capacity of HSBPs to bind HS arose through convergent evolution (20). Thus, HSBPs differ significantly from lectins, which share a limited number of structurally similar glycan-binding motifs or folds. The fact that HS binding can arise from many different protein folds may partly explain the wide variation in binding affinity between HS and HSBPs [dissociation constant (Kd) values generally between 1 nM and 10 μM]. It also fits with the general observations that most HSBPs can bind to more than one sequence in HS and that some binding sites in HS may interact with more than one protein. Apparently, this relationship, although somewhat promiscuous, is highly advantageous on the basis of evolutionary conservation of most HSBPs, ranging from ancient organisms to more evolutionarily modern species.
Main Categories of Heparan Sulfate–Binding Proteins
There are several major categories of HSBPs. These include chemokines and cytokines (~60); growth factors and morphogens that play essential roles in development and tissue repair (~50); blood coagulation factors such as serine proteases and their inhibitors (~25); extracellular structural proteins such as collagens, fibronectin, and vitronectin (~25); proteins involved in the complement pathways (~20); single-transmembrane signaling receptors (~15); and cell adhesion proteins (~10) (4). Other groups with a significant number of HSBPs (5–10 proteins) include the proteases in intracellular granules within the cells from the hematopoietic lineage, lipid-binding proteins involved in lipoprotein metabolism, and amyloid proteins. In addition, HS is widely exploited by pathogens for infection, and many pathogens have evolved or acquired HSBPs to assist their interaction with host cells (14).
Heparan Sulfate–Binding Proteins May Bind Other Glycosaminoglycans
In addition to HS, many cells express chondroitin sulfate (CS) and dermatan sulfate (DS), two other major types of sulfated GAGs. The structure of CS differs from that of HS in several ways, including the presence of different disaccharide repeats and glycosidic linkages (GlcAβ1–3GalNAcβ1–4 instead of GlcAβ1–4GlcNAcα1–4 in HS, where GalNAc refers to N-acetylgalactosamine), the lack of N-sulfation, a more even distribution of O-sulfation along the chain (usually at C4 or C6 of the GalNAc residues), and in general a lack of highly sulfated regions. DS has characteristics of both HS and CS in that its backbone is the same as that of CS, but a portion of its GlcA units undergoes epimerization to IdoA, which can then be sulfated at C2.
Given the structural similarity between CS, DS, and HS, it is not surprising that many HSBPs also bind these GAGs. Although the binding affinity to CS and DS is usually one-tenth to one-hundredth as strong as the binding affinity to HS, some HSBPs bind to DS with similar or even higher affinity than to HS (21–23). Therefore, it is wise to assess the binding property of a given HSBP to a full panel of GAGs. As a general rule, HSBPs that bind to a unique pattern of sulfated sugars tend to be highly selective for HS. A similar argument can be made for proteins that bind to DS, which also has sulfated domains containing IdoA (24). HSBPs that interact with HS simply through a charge-based mechanism tend to show low selectivity between HS, CS, and DS. This issue is addressed in greater detail in the following sections.
MODES OF HEPARAN SULFATE–PROTEIN INTERACTIONS
Basic Principles
Due to the highly anionic nature of HS, ionic interactions usually contribute a significant portion of the binding free energy (25–27). The negatively charged sulfate and carboxyl groups mediate interactions predominantly with positively charged lysine and arginine residues in the protein. Polar residues, usually asparagine, glutamine, and histidine, sometimes participate in hydrogen bonding with HS and can play critical roles in some HS–HSBP interactions. The nonionic contribution to binding free energy varies widely, ranging from less than 20% up to 70% (26–29).
Although electrostatics clearly play a major role in HS–protein interactions, note that these interactions do not occur randomly. This idea is supported by the fact that many proteins with high isoelectric points and with many lysine and arginine residues bind HS via a distinct subset of positively charged residues despite the presence of other positively charged surfaces. Furthermore, many HSBPs show selectivity between HS and similarly sulfated CS and DS chains, and some HS–protein interactions involve highly specific HS structures. In some cases, misplaced charges are detrimental to the interaction. Finally, if charge were the sole driver of HS–protein interactions, then the evolutionarily conserved, unsulfated NAc subdomains of HS would make little sense.
Binding of HSBPs to HS has many advantages. It enables spatial control of HSBPs by immobilizing and concentrating them at a specific location in the extracellular space; it improves the lifetime of HSBPs by protecting them from protease degradation and environmental damage; it allows multivalent interaction with HS and thus facilitates oligomerization; it promotes the binding efficiency of one HSBP to another HSBP when both bind to a single HS chain; and it functions as a switch to regulate the conformations of HSBPs. Therefore, gaining the capacity to bind HS creates a convenient and efficient way to adjust the form, location, and function of HSBPs.
Heparan Sulfate Can Act as a Tether
Perhaps the most fundamental activity associated with HS is its capacity to tether and present proteins at specific locations in tissues. HS is ideally suited for this role. First, the HS chains on cells can provide ~106 binding sites for ligands, a number that far exceeds the number of other types of receptors on the plasma membrane (30). Second, electrostatic interactions to a large extent drive the interaction between HSBPs and HS, which allows fast absorption of protein from the soluble phase (31, 32). Third, the length and flexibility of the HS chain enable bound HSBPs to sample the local region by unidirectional “sliding” along the chain or by mobility of the chain, allowing bound ligands to stay localized while sampling the adjacent area (33). Finally, binding to HS often lowers the susceptibility of ligands to proteolytic digestion, thereby increasing their life span in the extracellular environment (34, 35). We refer to such interactions as tethering because the interaction confines or limits the range of the bound ligand.
Tethering plays a critically important role in cell differentiation, a process that depends on gradients of morphogens and growth factors (36). Differentiation induced by the morphogens Wnt, hedgehog, and the bone morphogenetic proteins involves HS. The presence of only a few HS-deficient cells along the path of diffusion alters this process (37–40). How binding to HS affects the gradient is not fully resolved; it may involve not only effects on diffusion but also morphogen stability and endocytosis (41). Similarly, mice that express only the vascular endothelial growth factor (VEGF)-A121 isoform, which does not bind to HS, show gross defects in vascular patterning presumably because the isoform cannot form the necessary gradient to guide angiogenesis (42). Tethering also regulates axon pathfinding by facilitating gradients of axon guidance molecules (43). Tethering of chemokines via endothelial cell–surface HS generates so-called haptotactic gradients, which direct leukocyte trafficking to damaged or inflamed tissues and dendritic cells to lymph nodes (44, 45).
Diffusion of HSBPs is a dynamic process that involves initial binding, dissociation, and rebinding to HS chains. Recently, Duchesne et al. (33) examined how fibroblast growth factor 2 (FGF2) diffuses in a thin layer of extracellular matrix by using single-molecule tracking, providing the first clear picture of HS-dependent protein diffusion. While some molecules of FGF2 remain immobile or undergo restricted movement (range ~100 nm), others display either simple diffusive motion or directed diffusion ~15% of the time, with substantial translocation up to several micrometers even in fixed cells. This distance is far greater than the length of a single HS chain (≤200 nm); therefore, the translocation probably occurs by dissociation and retethering of FGF2 to nearby sites in different HS chains. Directed diffusion can then be explained by the spatial alignment of multiple HS chains that form a well-defined path of binding sites for FGF2. Tracking trajectories showed that two different FGF2 molecules actually travel the same path at different times. Different HSBPs display distinct binding kinetics for HS and bind to different subsequences; thus, several diffusion paths may coexist and form a network in the extracellular matrix or on the cell surface.
Heparan Sulfate–Induced Oligomerization
Oligomerization is one of nature’s fundamental ways of deriving new functionality from existing protein platforms (46). In general, compared with small proteins, large proteins can perform more complex biological activities because they often consist of multiple functional domains. However, errors occur more frequently during the assembly of multidomain proteins due to the complexity of the folding pathways (46). The assembly of large oligomeric proteins from small building blocks (monomers) circumvents this problem and contributes to the economy of the cell. In addition, oligomers can assemble and disassemble in a temporally and spatially specific manner, providing an additional layer of regulation to protein function.
Oligomerization, in particular homo-oligomerization, is highly prevalent in eukaryotic proteins (46). The factors that drive the formation of quaternary structures include hydrophobic and electrostatic interactions; hydrogen bonding; and van der Waal interactions, usually via a large oligomerization interface. Some secreted and membrane-anchored proteins find an alternative way to form oligomers by utilizing HS either as a bridge or as an allosteric factor. The examples described below illustrate these points.
FGF1.
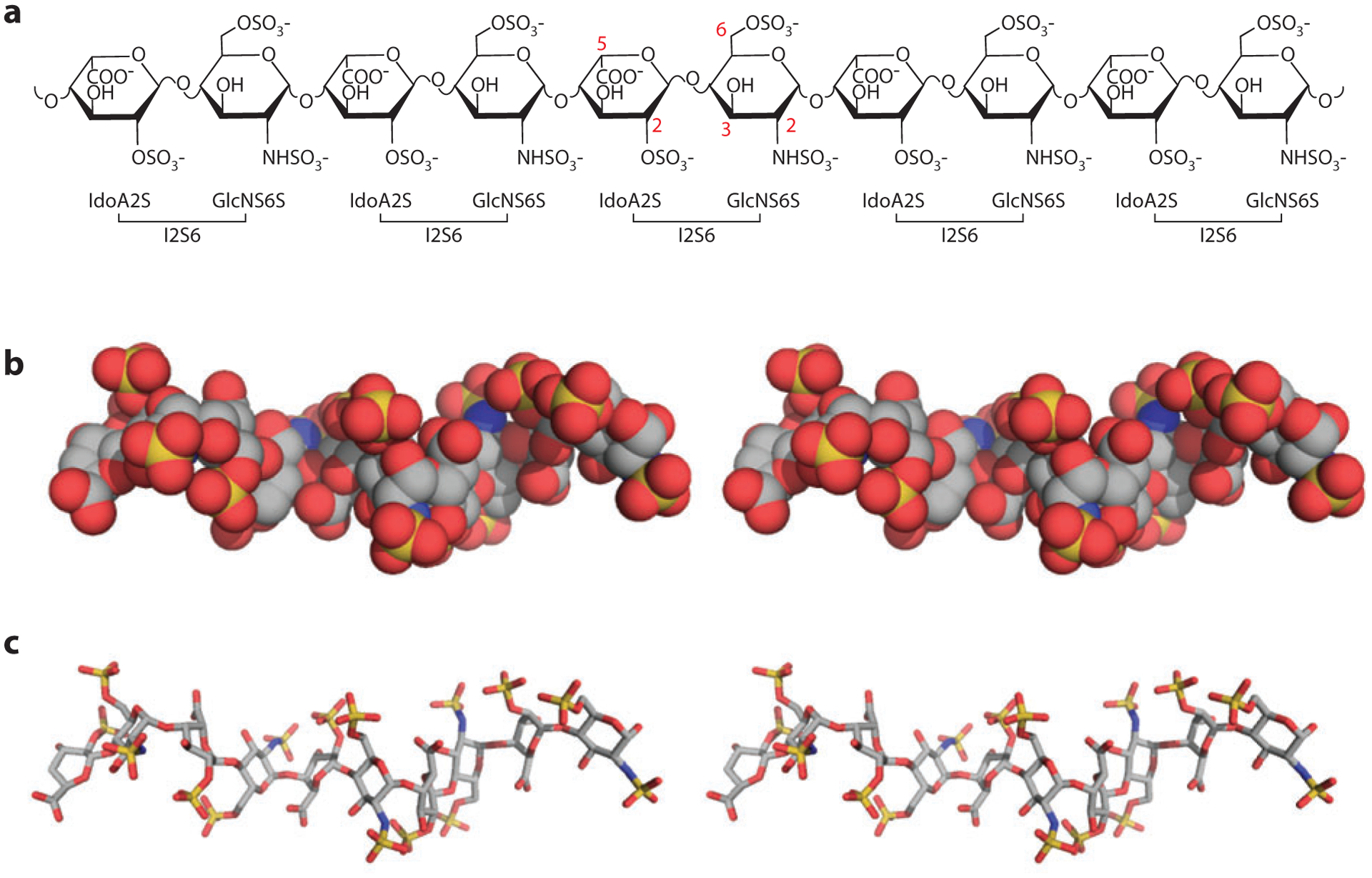
Many members of the FGF family interact with HS and form dimers (47, 48). The cocrystal structure of FGF1 and a heparin-derived decasaccharide [degree of polymerization (dp)10] revealed that protein–HS interactions drive dimerization in the absence of protein–protein interactions at the dimer interface (Figure 2a) (49). A recent isothermal calorimetry (ITC) study suggested positive cooperativity during FGF dimer formation, probably because binding of the first FGF1 monomer limits the conformational flexibility of the oligosaccharide and presents the preferred orientation of charged groups on the opposite side of the chain for binding to a second monomer (50). Importantly, the two FGF1 monomers bind HS asymmetrically due to the inherent asymmetry of the HS molecule (reducing and nonreducing ends). The same dimerization interface observed in the FGF1 dimer has been observed in two cocrystal structures of FGF1, FGF receptor 2 (FGFR2), and dodecasaccharides derived from heparin (51, 52). As discussed below, other crystal variants suggest that heparin oligosaccharides can also act as a template to oligomerize FGF with FGFR, independently of the formation of FGF dimers.
Figure 2.
Heparan sulfate (HS)–induced HS-binding protein oligomerization. In the three-dimensional drawings, one monomer is in green and one in pink; in the surface representations, positive electrostatic potential is in blue and negative electrostatic potential is in red. For the oligosaccharide, carbon is gray, oxygen red, nitrogen blue, and sulfate yellow. (a) Structure of FGF1 (fibroblast growth factor 1) dimer and bound oligosaccharide [Protein Data Bank (PDB) identifier 1AXM]. (b) Structure of the dimeric V-C1 domains of RAGE (receptor for advanced glycation end products) (PDB 4IM8). The dodecasaccharide is manually modeled into the structure on the basis of the observed partial electron density. (c) Structure of the dimeric E2 domain of amyloid precursor–like protein 1 (APLP-1) and bound oligosaccharide (PDB 3QMK). (d) Structure of dimeric interleukin-8 (PDB 2IL8) and a modeled oligosaccharide (degree of polymerization: 20). (e) Structure of dimeric CXCL12 (PDB 2NWG) and a modeled decasaccharide.
RAGE.
The receptor for advanced glycation end products (RAGE) becomes activated in many inflammatory conditions via ligands such as S100 proteins, high-mobility group protein B1, and other damage-associated molecular pattern proteins (DAMPs) released from activated cells or necrotic tissue. Oligomerization of RAGE is essential for signal transduction, which depends on engagement with ≥dp12 oligosaccharides. Binding to heparin or HS precedes ligand binding and presumably assembles RAGE into a functionally ready state for signaling prior to engagement of its cognate protein ligands. ITC experiments demonstrated a 2:1 stoichiometry for RAGE V-C1 subunits to oligosaccharide. The crystal structure of the RAGE hexamer obtained in the presence of heparin-derived dodecasaccharide demonstrates a trimer of dimers in which each dimer was stabilized by one dodecasaccharide bound at docking sites that were defined by mutagenesis (Figure 2b) (53). In addition, small-angle X-ray scattering indicates that the hexamer structure is retained in solution. The RAGE dimer has a small hydrophobic dimerization interface next to the HS-binding site with a buried surface area of ~300 Å2. In spite of its small size (too small to support a stable dimer by itself under normal conditions), the hydrophobic interface is essential for the formation of stable dimers. The hydrophobic interactions at the protein–protein interface and the electrostatic interactions mediated by HS are energetically coupled to promote RAGE dimerization. Importantly, recent data demonstrate that RAGE oligomerization induced by HS is required for signaling (53, 54).
Amyloid precursor protein.
Amyloid precursor protein (APP) is the precursor for amyloid-β, which accumulates in Alzheimer disease (55). APP and its homologs amyloid precursor–like proteins 1 and 2 (APLP-1 and APLP-2) are single-transmembrane proteins that play important (but not fully understood) functions in synapse organization (56–58). All APP family proteins bind HS, which is essential for APP dimerization at the cell surface (59, 60). APP is unusual in that it possesses two HS-binding sites in two different domains, E1 and E2 (61). Apparently, both domains can bind HS independently with micromolar association constants (62, 63). Quite remarkably, both E1 and E2 undergo HS-dependent dimerization with a 2:1 stoichiometry of protein:oligosaccharide (64, 65). The interaction between HS and the higher-affinity HS-binding domain E2 was characterized by cocrystallization (65). Similar to the RAGE dimer, the HS-binding site is located next to the dimerization interface (Figure 2c). Although the hydrophobic dimerization interface of the E2 domain (1,400 Å2) is much larger than that of RAGE, the E2 domain remains predominantly monomeric in solution (Kd for monomer–dimer equilibrium = 161 μM) (62). Clustering of positively charged residues near the dimerization interface presumably generates electrostatic repulsion and thus destabilizes the dimer. These positive charges, many of which are part of the HS-binding site, are effectively neutralized by bound HS, which consequently shifts the equilibrium toward dimer formation (Kd = 7.8 μM in the presence of heparin) (62).
Chemokines.
With very few exceptions, all chemokines have a strong tendency to oligomerize, and oligomerization plays an essential role in chemokine biology (44, 66, 67). Most chemokines exist as either monomers or dimers at physiological concentrations, but their interaction with HS drives the equilibrium toward the formation of dimers or higher-order oligomers (67, 68). The overall folds of most chemokines are very similar (a long N-terminal loop followed by a 310 helix, a β-sheet formed by three β-strands ending with a C-terminal α-helix). However, the mechanism of oligomerization and HS binding varies for different chemokines (69). Many chemokines in the CXCL family have a dimer interface formed by the first of the three β-strands. However, the HS-binding sites can differ significantly within this family. The HS-binding sites of interleukin (IL)-8 and platelet factor 4 (PF4, or CXCL4) are located on opposite sides of the dimerization interface (Figure 2d) (70, 71), whereas the HS-binding site of stromal cell–derived factor 1 (SDF-1, or CXCL12) is in close proximity to and just above the dimer interface (Figure 2e) (72). In contrast, most CCL chemokines use a different dimerization interface that involves interaction of largely unstructured N-terminal loops (73). The HS-binding site of this family is also adjacent to the dimerization interface. Interestingly, when a CXCL chemokine (such as PF4) and a CCL chemokine, monocyte chemoattractant protein 1 (MCP-1, or CCL2), form tetramers, both dimerization interfaces are used (71, 73). In these cases, the HS-binding sites from all four subunits form a ring of positive charges that surrounds the whole tetramer. The physiological significance of chemokine oligomerization is not yet fully understood. The most likely role of chemokine oligomerization involves better retention at the luminal endothelial surface because of the higher affinity of oligomers for HS, better stability and longer half-life because of protection of oligomers from proteolytic digestion, and an added layer of regulation based on monomer–oligomer equilibrium (67).
Other HSBPs that depend on HS for oligomerization include FGF2 (74), thrombospondin (75), hepatocyte growth factor/scatter factor (HGF/SF) (76), the membrane proteins neuropilin-1 and -2 (77), and the receptor protein tyrosine phosphatase σ (RPTP-σ) (78). Many other as-yetuncharacterized HSBPs are also likely to undergo HS-induced oligomerization.
Heparan Sulfate as a Scaffold for Protein–Protein Interactions
It is well known that so-called scaffold proteins play crucial roles in orchestrating signal transduction events (79). The principal role of a scaffold protein is to bring two proteins into close proximity to provide a greater chance of successful engagement. Scaffold proteins not only allow more productive binding between the interacting partners but also afford precise regulation of where and when signaling events initiate. Structural features of HS, such as the length, flexibility, and structural diversity along the chain, make HS an ideal scaffold molecule. Indeed, a common characteristic of HS is that it can act as a scaffold to promote protein interactions. In the following subsections, we describe two well-established systems in which HS serves as a molecular scaffold: serine protease–serpin interactions and ligand–receptor interactions.
Antithrombin/thrombin.
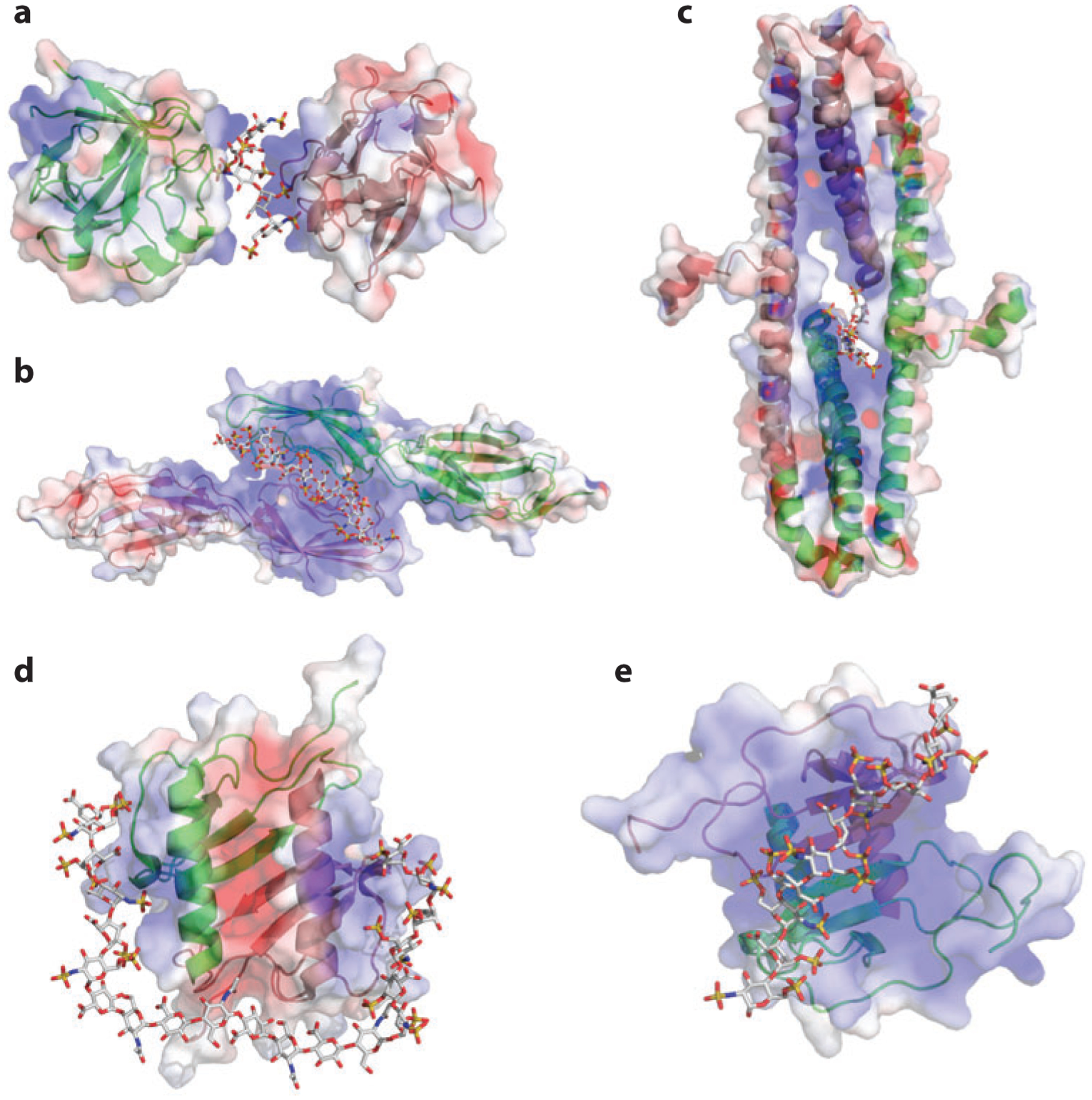
Thrombin occupies a prominent position in the blood coagulation cascade because it directly converts fibrinogen into fibrin, the major component of blood clots (80). Several serine protease inhibitors termed serpins regulate the enzymatic activity of thrombin; AT is the best-known member of this family (81). In the absence of HS, AT is not an efficient inhibitor of thrombin; however, its inhibitory rate constant increases more than three orders of magnitude when HS is present (82). The underlying reason for this dramatic rate enhancement is HS’s ability to act as a scaffold to approximate AT and thrombin, both of which are HSBPs (83). By binding simultaneously to both enzyme and substrate, HS greatly increases the chance of successful engagement of thrombin and the flexible reactive central loop (RCL) of AT (84). Interestingly, investigators initially observed that oligosaccharides of ≥dp18 are required for maximal rate enhancement (85). However, only an appropriately sulfated pentasaccharide suffices for AT binding, and a sulfated hexasaccharide is adequate for thrombin binding (86, 87). Two cocrystal structures of the ternary complex of AT, thrombin, and a synthetic HS mimic (84, 88) provided the explanation for the requirement of an oligosaccharide of ≥dp18. The distal locations of the HS-binding sites of AT and thrombin require an arrangement of two NS domains with an intervening inert linker of dp8 (Figure 3a). This arrangement is reminiscent of many scaffold proteins that are organized such that distinct protein-binding domains are separated spatially by an extended disordered region (79).
Figure 3.
Heparan sulfate (HS) acts as a molecular scaffold. In all structures, proteins are shown in surface electrostatic potential, and the oligosaccharides are shown in stick representation. Each HS-binding site is enclosed in a dashed circle. (a) Cocrystal structure of thrombin, antithrombin (AT), and a hexadescasaccharide heparin mimic [Protein Data Bank (PDB) identifier 1TB6]. (b) Cocrystal structure of thrombin, protein C inhibitor (PCI), and a heparin-derived tetradecasaccharide. Because the tetradecasaccharide cannot be fully resolved in the cocrystal structure except for two sugar residues, it is manually modeled here (PDB 3B9F). (c) A model of the ternary complex of activated protein C (APC), PCI, and tetradecasaccharide based on PDB 3B9F and biochemical evidence (92). (d) Cocrystal structure of fibroblast growth factor 1 (FGF1), FGF receptor 2 (FGFR2, D2 and D3 domains), and heparin-derived decasaccharide (PDB 1E0O). (e) Cocrystal structure of FGF2, FGFR1 (D2 and D3 domains), and heparin-derived decasaccharide (PDB 1FQ9).
Protein C inhibitor/thrombin.
Protein C inhibitor (PCI) is another serpin that acts on thrombin. Although the structure of PCI is similar to that of other serpins, including AT, the HS-binding site is located in a completely different region (89). The HS-binding site of PCI is situated much closer to the PCI/thrombin interface than that found in AT/thrombin complexes, enabling the formation of a composite HS-binding site composed of domains from both PCI and thrombin (Figure 3b) (90). The close proximity between the two HS-binding sites enables shorter but fully sulfated oligosaccharides (≥dp14) to act as a scaffold, in contrast to the dp18 oligosaccharide required for bridging AT and thrombin. This arrangement suggests that a single NS domain may suffice to facilitate the formation of a PCI/thrombin complex.
PCI actually has procoagulant activity by inhibiting activated protein C (APC), another serine protease involved in coagulation (81). PCI inhibition of APC also requires HS to form a ternary complex (91). Interestingly, the HS-binding site of APC is distinct from the one in thrombin and is located on the opposite side of the molecule (28), which poses a steric problem for binding to HS (Figure 3c). The HS-binding site of PCI has substantial plasticity, which allows HS to bind in a quite different orientation to bridge the PCI/APC complex, in contrast to the orientation observed in the PCI/thrombin complex (92). What emerges from these various studies is HS’s enormous versatility as a scaffold.
FGF/FGFR.
The interactions between FGFs and their receptors have been extensively studied and crystal structures of eight different FGF/FGFR complexes have been solved over the years (47). All FGFR isoforms bind HS with moderate to high affinity (93–95). FGF and FGFR can form a complex in solution, but these complexes tend to dissociate during size-exclusion chromatography (96, 97). However, if heparin is included in the mixture, the FGF/FGFR/heparin ternary complex is much more stable and can easily be purified by chromatography (97). Interestingly, two cocrystal structures revealed two different ways that HS interacts with the FGF/FGFR complex that vary in stoichiometry and orientation of the subunits; these data may reflect different purification methods and crystallization conditions (Figure 3d,e). Despite the differences, both crystal structures showed that the HS-binding site of FGFR, which is located in the D2 domain, merges with the HS-binding site of FGF to form a large composite docking site for HS (Figure 3d,e). Although the exact orientation differs, such composite HS-binding sites are observed in virtually all FGF/FGFR combinations (51, 52, 96, 98). The bridging function of HS, mediated by a relatively short, highly sulfated oligosaccharide sequence, resembles the arrangement of HS in the PCI/thrombin complex. This mode of scaffolding might be relevant to other ligand–receptor interactions, including those between Slit and Robo (99) and between VEGF and neuropilin (77).
Heparan Sulfate as an Allosteric Regulator
All proteins are potentially allosteric. Simply put, allostery is the effect of change at one site on the activity at another site (100). Proteins exist in equilibrium in an ensemble of conformations. Thus, the allosteric regulation of protein is essentially a redistribution of the preexisting conformations upon effector binding that biases the equilibrium toward a certain conformation. If the selected conformation favors interaction with another molecule, positive cooperativity occurs; if the selected conformation disfavors interaction with another molecule, negative cooperativity occurs. Importantly, allostery does not necessarily involve conformational changes of the target-binding site; it can be transmitted simply by changes in protein dynamics (101).
HS generally is not considered a signal-inducing ligand or substrate for HSBPs, because binding itself neither evokes signal transduction nor generates products (with the exception of HS biosynthetic and degradation enzymes). Therefore, HS can best be viewed as an effector molecule, namely an allosteric modulator of HSBPs. So far, two HSBPs are known to show HS-dependent conformational change on the basis of evidence from crystallization studies; these HSBPs are AT and a viral protein known as Vaccinia virus complement control protein (VCP).
Antithrombin.
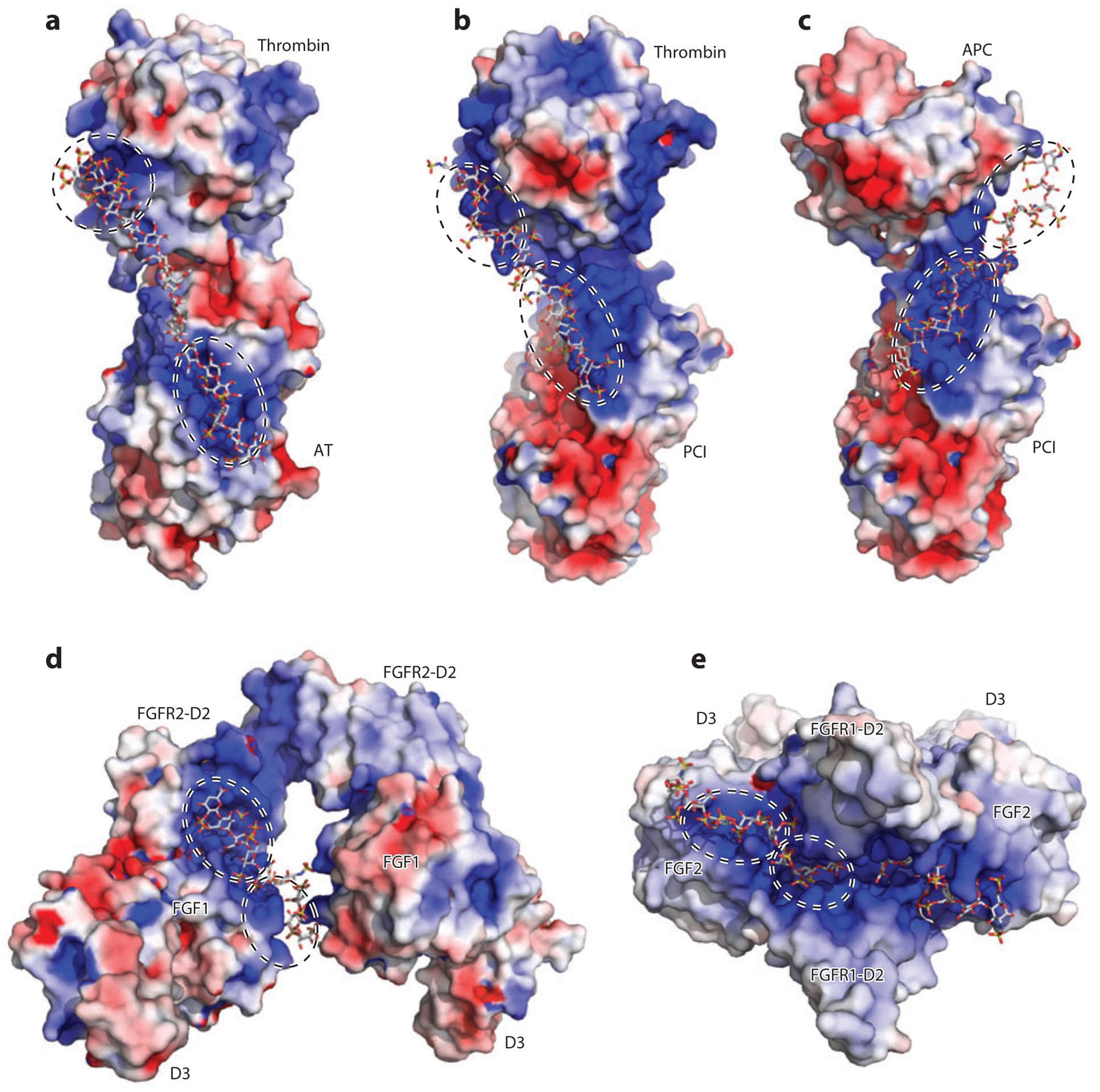
All serpins have an RCL that acts as bait to achieve inhibition of target serine protease (81). AT is unique among serpins in that its RCL adopts a more constrained conformation, which makes it less accessible for certain proteases (Factors Xa and IXa) and is responsible for the poor inhibitory activity of native AT (102). However, upon binding of a unique pentasaccharide sequence containing a rare 3-O-sulfated glucosamine, the RCL constraint is removed through an allosteric mechanism that is now well understood (102, 103). The conformation change starts with the elongation of helix D at its C terminus upon pentasaccharide binding, which pushes the first two β-strands toward the center of the four-strand A-sheet; in turn, these β-strands squeeze out the N-terminal portion of the RCL to allow it to adopt a more flexible conformation (Figure 4a). This flexible conformation enables two critical electrostatic interactions between Factor Xa (as well as Factor IXa) and AT (86, 104). The HS-binding site of AT plays a pivotal role in triggering the conformational change. Comparison between the pentasaccharide-bound and native AT structures reveals the key structural elements of the HS-binding site that undergo significant rearrangement and allow the side chains of the HS-binding amino acid residues to move by 5 to 17 Åto accommodate the pentasaccharide (Figure 4b) (103).
Figure 4.
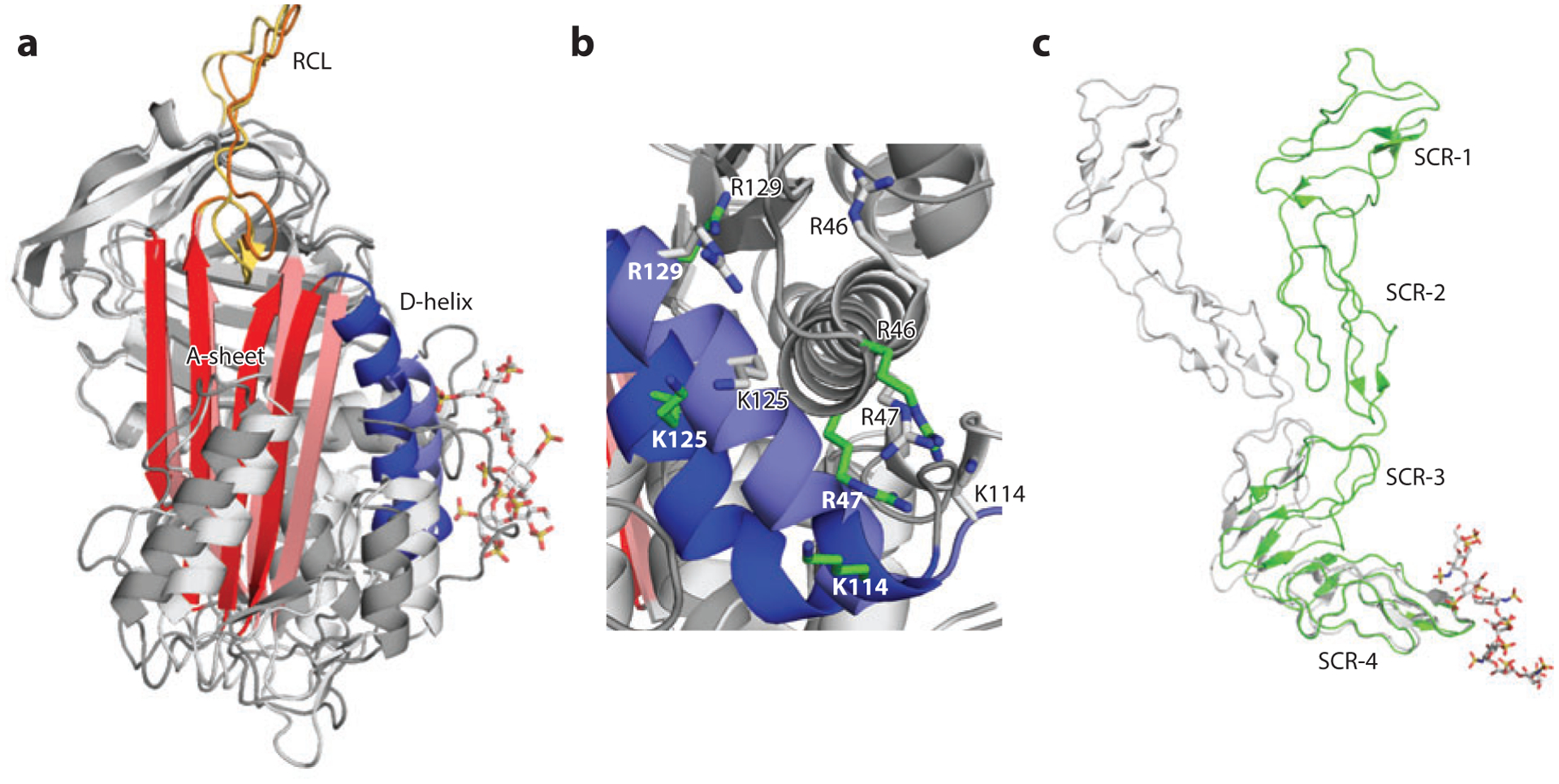
Heparan sulfate (HS) acts as an allosteric regulator. (a) Overlay of native antithrombin [Protein Data Bank (PDB) identifier 1E04) (light gray) and pentasaccharide-bound antithrombin (PDB 1AZX) (dark gray). The important structural elements are the D-helix in purple (native) and blue (bound), the A-sheet in pink (native) and red (bound), and the reactive central loop (RCL) in yellow (native) and orange (bound). (b) Rearrangement of HS-binding residues following pentasaccharide binding. For clarity, the pentasaccharide is not displayed. The native conformation of HS-binding residues is shown as gray sticks, and the pentasaccharide-bound conformation of HS-binding residues is shown as green sticks. (c) Overlay of native Vaccinia virus complement control protein (VCP) (PDB 1G40) (gray) and decasaccharide-bound VCP (PDB 1RID) (green). Abbreviation: SCR, short consensus repeat.
VCP.
VCP is a viral HSBP that helps Vaccinia virus evade host defense by downregulating complement pathways (105). This protein is particularly interesting to immunologists because it bears high structural homology to two host proteins that regulate complement activation, Factor H (FH) and C4b-binding protein, both of which bind to HS (106, 107). FH and C4b-binding protein play essential roles in distinguishing host and pathogen by downregulating complement pathways at the host cell surface. A cocrystal structure of VCP and heparin decasaccharide revealed the unexpected finding that HS binding to the C-terminal short consensus repeat (SCR) domain causes a significant conformational change that involves domain movement (Figure 4c) (107). In its native conformation, the last two SCRs of VCP, SCR-3 and SCR-4, form an angle of ~120° (108). When HS binds to the tip of SCR-4, the angle between SCR-3 and SCR-4 declines to ~80°, bringing SCR-3 much closer to SCR-4. HS may selectively bind and stabilize this closed conformation (107). Because SCR-4 contains several residues that interact with complement C3b (107), the changes in its relative orientation to SCR-1 and SCR-2 (which move with SCR-3 as a rigid body) would alter C3b-binding affinity and kinetics. HS promotes binding of FH to C3b in solution, probably by stabilizing the closed conformation of SCR domains of FH (109). Thus, HS appears to induce positive cooperativity toward C3b binding.
Interestingly, the vast majority of HSBPs do not show any noticeable backbone conformational changes upon HS binding on the basis of available structures of the bound and unbound proteins. However, the lack of observable conformational changes in crystals does not mean they do not occur, because crystallization inevitably biases proteins toward conformations that are more conducive to crystal formation and therefore may obscure conformational changes that would occur in solution. Also, as stated above, a conformational change at the target-binding site is not required for HS to have an allosteric effect. Presumably, HS binding can have a significant effect on the dynamic fluctuations in the structure of HSBPs and thus affect their interaction with a second molecule at a distal site. The most direct way to test this hypothesis experimentally is to use ITC. In one study, Brown et al. (50) examined FGF1 binding to FGFR2 in the presence or absence of heparin-derived hexasaccharide. Although the binding of hexasaccharide did not cause any conformation change of FGF1, the binding affinity of the hexasaccharide/FGF1 complex to FGFR2 (0.3 μM) was ~14-fold higher than the affinity between FGF1 and FGFR2 (4.3 μM), suggesting that the hexasaccharide induced positive cooperativity.
SPECIFICITY OF HEPARAN SULFATE–PROTEIN INTERACTIONS
Specific or Nonspecific?
A central question is whether the interaction between HS and an HSBP is specific; that is, whether a specific sequence of HS, defined by the arrangement of sulfated disaccharides and IdoA, drives the interaction or if a number of sequences behave equivalently (110, 111). During the past two decades, many biochemical and biophysical studies have revealed that specificity is manifested at many different levels. For some proteins, such as AT and FGF2, very distinct modifications of HS are required for optimal interaction (modification-specific interactions) (112, 113), whereas for other proteins, such as IL-8 and interferon (IFN)-γ, the specificity resides in the domain structure of HS rather than relying on specific modifications (domain-specific interactions) (114, 115). For still other proteins, such as thrombin, the interaction appears to be nonspecific and dependent solely on charge density (charge-based interactions) (27). Importantly, specificity in binding should not be confused with biological significance, because even a nonspecific interaction (such as HS–thrombin) has a profound physiological consequence. The observation that increases of sulfation at some sites can compensate for alterations in sulfation at other positions in some developmental contexts supports this idea (116). Lastly, our understanding of the sequence specificity for many HSBPs is undoubtedly incomplete because of a lack of structurally defined oligosaccharides to probe the interaction. In many cases, we know only that certain sets of sulfate groups are important, but we cannot define a specific arrangement of sulfated disaccharides.
Modification-specific interactions.
As mentioned in the Introduction, each disaccharide unit can be modified at five different positions, which could give rise to up to 48 different disaccharides; however, only ~20 are commonly observed in HS (10). With this restriction, the number of permutations resembles the number of primary amino acid sequences found in proteins (21 amino acids). This observation speaks to the enormous coding capacity of HS, which prompted many researchers to hypothesize that distinct combinations of disaccharides, namely HS sequences, represent a code and dictate the specificity of HS–HSBP interactions (43). Because the term code can imply a template-driven process similar to that involved in transcription/translation, we prefer not to use this terminology but rather focus on specificity and selectivity based on structure.
The well-characterized HS–AT interaction supports this hypothesis. The high-affinity interaction between AT and HS requires a rare 3-sulfo-N-sulfoglucosamine residue (GlcNS3S) in a very specific context. Binding studies suggested that the 3-O-sulfate group contributes ~60% of the binding free energy and that removal of this modification causes a 105-fold reduction in binding affinity to AT (112). Mechanistic studies determined that the 3-O-sulfate group, along with its amino acid partner in AT (Lys114), occupies a pivotal position in organizing both ionic and nonionic interaction networks that are responsible for the AT–HS interaction (Figure 5a). The surrounding sugar residues also play important roles in promoting this interaction. For example, there must be a nonsulfated glucuronic acid at the nonreducing side of the GlcNS3S unit (Figure 5a). If this residue is replaced with IdoA or 2-sulfo-IdoA (IdoA2S), then the oligosaccharide shows greatly reduced binding to AT (118). Although only a few proteins are known to depend on 3-O-sulfation, there will probably be many others because of the large number of HS 3-O-sulfotransferases (10, 119). Whether these proteins exhibit high selectivity, similarly to AT, awaits their identification and characterization.
Figure 5.
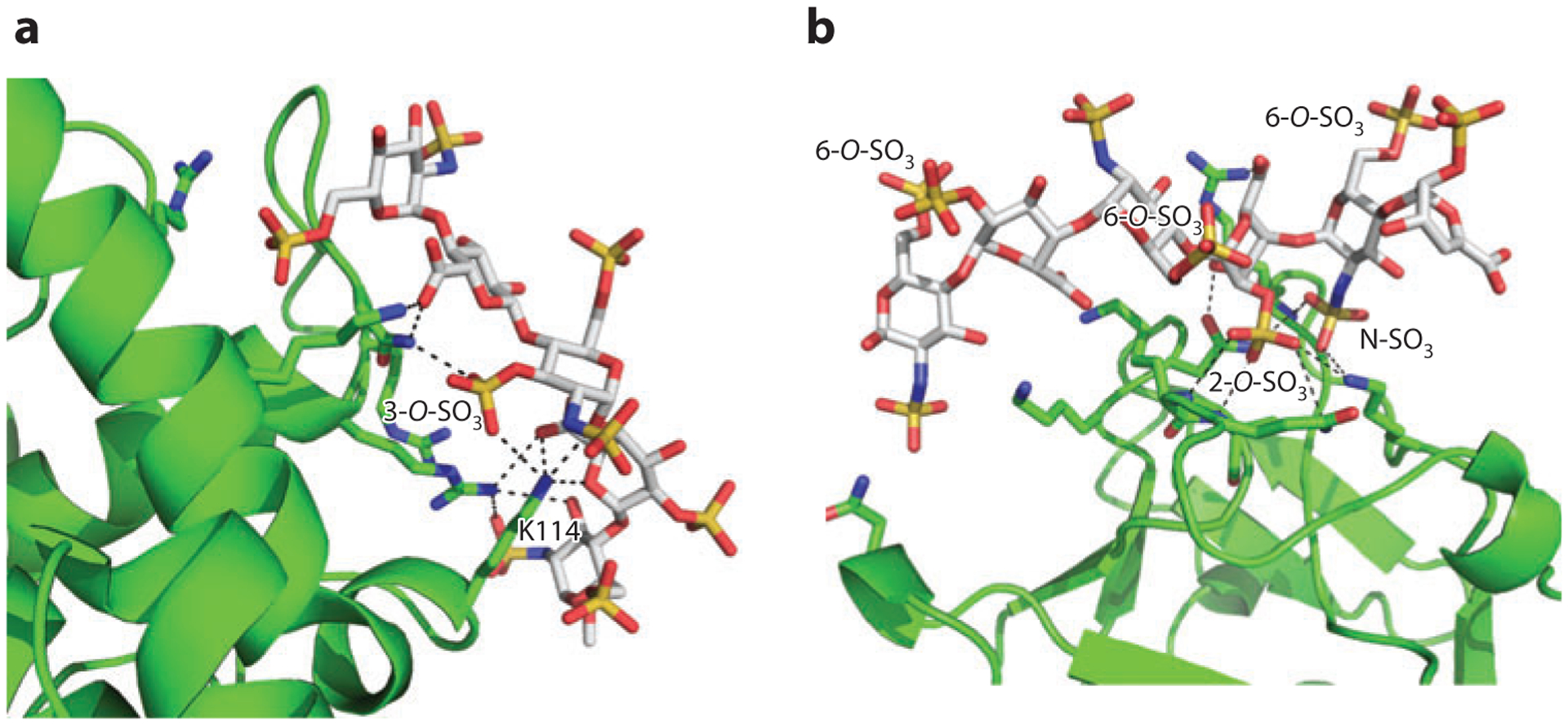
Structural basis of modification-specific heparan sulfate (HS)–HS-binding protein (HSBP) interactions. (a) Pentasaccharide–antithrombin (AT) interaction [Protein Data Bank (PDB) identifier 2GD4]. HS-binding residues of AT are shown as green sticks, and the pentasaccharide is shown as gray sticks with oxygen in red, nitrogen in blue, and sulfate in yellow. The ionic and nonionic interactions stabilized by 3-O-sulfate and K114 (both directly and indirectly) are shown as dashed lines. (b) Hexasaccharide–fibroblast growth factor 2 (FGF2) interaction (PDB 1BFC). HS-binding residues and hexasaccharide are colored as in panel a. The ionic and nonionic interactions that involve the critical disaccharide—IdoA2S-GlcNS6S—are shown as dashed lines.
FGF2 is another well-characterized HSBP. Formation of FGF2/HS complexes depends critically on IdoA2S and N-sulfated glucosamine (GlcNS) (113, 120). The cocrystal structures of FGF2 and hexasaccharide show that the major interaction occurs via one disaccharide that makes contact with the so-called high-affinity HS-binding site of FGF2 (121). This disaccharide makes nine salt bridges and hydrogen bonds with FGF2; of these nine, six are contributed by the IdoA2S residue and three by the N-sulfate group, which explains the importance of these two modifications (Figure 5b). In contrast, the three 6-O-sulfate groups of the hexasaccharide do not contact FGF2. The crucial role of IdoA2S and GlcNS in FGF2/HS interaction is further confirmed by the cocrystal structure of FGF2/FGFR1/decasaccharide, in which 8 out of 10 hydrogen bonds between FGF2 and decasaccharide involve these two sugar residues (96). Interestingly, in this structure 6-O-sulfates contribute two hydrogen bonds with FGF2, suggesting that a certain degree of flexibility is allowed in the interaction. Although 6-O-sulfate groups may be dispensable for interacting with FGF2, they are indispensable for promoting FGF2/FGFR signaling due to their extended hydrogen bonding with FGFR (122, 123).
A survey of several heparin oligosaccharide/HSBP complexes shows that many sulfate groups in the oligosaccharides do not contact the protein; therefore, most probably do not contribute to the binding free energy. Apparently, many HSBPs tolerate these unbound negative charges, which would explain the successful use of highly sulfated heparin-derived oligosaccharides in many studies. However, the use of highly sulfated heparin can be problematic. One study, which tested the binding specificity of FGF7 by using heparin-derived octasaccharides, found that moderately sulfated octasaccharides with 7 to 8 sulfates showed higher affinity to FGF7 than did fully sulfated octasaccharides with 11 to 12 sulfates (124). Thus, fully or nearly fully sulfated sequences (such as the commonly used heparin-derived oligosaccharide) may create artifacts and potentially obscure a binding site in less-sulfated domains. In other words, the natural sequences presented in HS may be more selective than the fully sulfated sequence in heparin. Recent advances in chemical and chemoenzymatic synthesis of structural defined oligosaccharides should provide the reagents needed to decode the relevant binding sequences in HS (125).
Domain-specific interactions.
A distinct structural feature of HS is that it is arranged as alternating sulfated NS domains, rarely sulfated NAc domains, and mixed NAc/NS domains (10). Analyses by heparin lyase and nitrous digestion suggest that the length of NS and NAc domains varies considerably, with NS domains that range from dp6 to dp16, NAc domains as large as dp18, and NAc/NS domains that account for as much as one-quarter of the length of the chain (8, 9, 126). Investigators initially assumed that proteins interacted with one sulfated NS domain, which is the case for many HSBPs. However, later research showed that many proteins, mostly in the chemokine and cytokine families, bind to two adjacent NS domains simultaneously. A few well-characterized examples include IL-8, PF4, MIP-1α, and IFN-γ (114, 115, 127, 128).
Under physiological conditions, IL-8 binds to oligosaccharides of at least dp18. Detailed structural analyses of the bound HS suggest that the active oligosaccharide contains two dp4–dp6 NS domains at the ends and one intervening NAc domain (up to dp14) (115). Each NS domain binds to one subunit of the IL-8 dimer. PF4, which binds HS as a tetramer, requires a much larger HS fragment for interaction (dp42), with a domain structure resembling the bipartite structure of the IL-8-binding fragment. The dp42 fragment consists of one dp10–dp14 or two dp6–dp8 NS domains at each end and one dp14–16 NAc domain (127). The HS-binding sequence of the MIP-1α dimer is also large (~dp34) and is composed of two long NS domains (dp12–dp14) at both ends and a relatively small NAc domain (dp8) (128). This arrangement is almost the opposite of that observed for IL-8. Finally, the preferred binding sequence of the IFN-γ dimer is ~dp46 (114) and is organized as two terminal NS domains of typical size (dp6–dp8) and an unusually long intervening NAc domain with a length of dp32. The utility of such a long linker, which is commonly regarded as an inert structure that has no interaction with protein, remains obscure.
A requirement for bipartite binding domains in HS may make suitable sites for binding somewhat rare. A recent study demonstrated that changing the domain structure of HS by inactivation of Hs2st (which causes an increase in glucosamine N-sulfation and 6-O-sulfation by an unknown compensatory mechanism) increased by 10-fold the number of binding sites for IL-8 on endothelial HS and increased acute inflammatory responses in mice (129). This finding raises the possibility that regulation of N-sulfation could regulate the capacity of HS to present chemokines and cytokines. Some data suggest that inflammation alters the expression of different enzymes in the pathway, but whether these changes alter the binding of chemokines is unknown (130, 131).
Charge-based interactions.
Many HSBPs interact with HS in a more flexible manner. Thrombin has often been cited as a classical example of a nonspecific charge-based interaction partner of HS. Early binding studies revealed that the thrombin–HS interaction showed a strong dependence on salt concentration, showed a low dependence on nonionic interactions, and demonstrated an increase in apparent binding affinity with increasing length of oligosaccharide (27). These characteristics contrast with the behavior of AT–HS and FGF2–HS interactions, which show mild dependence on salt concentration, have large nonionic contributions to binding, and have a binding affinity that is relatively indifferent to the size of the oligosaccharide (29, 112). The cocrystal structures of thrombin and a heparin octasaccharide showed opposite orientations for the oligosaccharide. Although the amino acids involved in binding remain the same, any given residue can bind to different sulfate groups in the two orientations. For instance, residue Lys236, which contributes the most to the binding free energy, interacts with N-sulfate and carboxylate groups in one structure, whereas in another structure, it interacts with 2-O-sufate and a different carboxylate group. Thus, the arrangement of positively charged residues of the thrombin–HS–binding site enables it to tolerate different negative-charge patterns in HS, as long as the interaction satisfies the required five to six ionic interactions (87).
HGF/SF is a large growth factor that relies on HS for activation of its cognate receptor, the tyrosine kinase MET. The extent of HGF/SF–HS interaction strongly correlates with the overall sulfation of the chain, but the interaction does not depend on the specific location of the sulfate groups; selectively de-N-, de-2-O-, and de-6-O-sulfated heparins bind HGF/SF equally well (132). Remarkably, more extensively desulfated heparin lacking both N- and 2-O-sulfates or both 2-O- and 6-O-sulfates can associate with HGF/SF in gel mobility shift assays and promote HGF/SF signaling (132). Cocrystal structures of HGF/SF/decasaccharide complexes show that several nonidentical decasaccharide binding modes exist. Among the seven amino acid residues that form the HS-binding site, only three (Thr61, Lys63, and Arg73) make invariant contacts with HS in all binding modes, whereas interactions with the other four basic residues are more variable. The nonspecific nature of the HS-binding site of HGF/SF also explains its capacity to interact with DS with an affinity comparable to HS (132).
Note that this type of charge-based, nonspecific interaction does not imply low affinity binding or lack of biological impact. It should be considered simply as another way that some HSBPs can exploit the structure of HS. Low selectivity can also ensure binding across different cell types and HS presentations, which could prove beneficial for housekeeping proteins, DAMPs, and receptors for viral or microbial infection. Also, a general feature of this type of HSPG–HS interaction is that binding strength increases with increasing level of sulfation. This observation has prompted some researchers to hypothesize that the purpose of graded levels of sulfation of HS chains is to enable a so-called modulated functional response, which may well be one of the most fundamental ways in which HS regulates various biological systems (111).
Structural Features of Heparan Sulfate–Binding Sites
As discussed above, HSBPs consist of proteins that interact with HS by way of many different protein folds. In this section, we describe well-characterized HSBPs and identify the structural features that are common to their HS-binding sites, despite their overall structural dissimilarity. The structures of more than 20 HSBPs are known on the basis of crystal or NMR structures (either in apo form or in complex with an oligosaccharide). Table 2 lists the residues and secondary structure elements in their HS-binding sites, the dimensions of the HS-binding sites, and the dissociation constants of binding to heparin.
Table 2.
Structural features of selected heparan sulfate–binding sites
| HSBP | Basic residues | Polar residuesa | Dimensionsb | Secondary elementsc | Affinity for heparin (Kd)d |
|---|---|---|---|---|---|
| FGF1 | K112, K113, K118, R119, R122, K128 | N18, N114, Q127 | 600 Å2, dimer (12 × 25 Å × 2) | Loop, loop, loop | 50–140 nM |
| FGF2 | K27, R121, K126, K136 | N28, N102, Q135 | 300 Å2 (12 × 25 Å) | Loop, loop, loop | 30–50 nM |
| HGF/SF | K58, K60, K62, R73, R76, K78 | T61 | 300 Å2 (12 × 25 Å) | Loop, strand, helix, loop | 1–3 nM |
| VEGF | R123, R124, R156, R159 | ND | 450 Å2, dimer (15 × 15 Å × 2) | Loop, loop | 100 nM |
| HMGB1 | K87, K88, K90, K96, R97, K150 | ND | 420 Å2 (15 × 28 Å) | Loop, helix | 5 nM |
| Cyclophilin B | K4, K5, K6, K9, K35, K97, K99, K182 | ND | 320 Å2 (16 × 20 Å) | Loop, loop, loop, loop | 16 nM |
| IL-8 (CXCL8) | K20, R60, K64, K67, R68 | ND | 430 Å2, dimer (12 × 18 Å × 2) | Loop, helix | 2 μM |
| PF4 (CXCL8) | R20, R22, K61, K62, K65, K66 | ND | 880 Å2, tetramer (10 × 22 Å × 4) | Loop, helix | 16 nM |
| SDF-1 (CXCL12) | R20, K24, K27, R41, K43, R47 | ND | 520 Å2, dimer (10 × 52 Å) | Loop, strand, strand, loop | 30 nM |
| MCP-1 (CCL2) | R18, K19, R24, K49, K58 | H66 | 1150 Å2, tetramer (12 × 24 Å × 4) | Loop, loop, loop, helix | 1.5 μM |
| FGFR1 | K160, K163, K172, K175, K177, K207, R209 | None | 700 Å2, dimer (16 × 22 Å × 2) | Loop, loop, strand, strand | 3 μM |
| Nrp-1 | R359, K373, K509, R513, K514, K516 | ND | 480 Å(12 × 40 Å) | Strand, loop, loop | 30 nM |
| RAGE | K39, K43, K44, R104, K107, R216, R218 | ND | 1050 Å, dimer (19 × 55 Å) | Loop, loop, loop | 3 nM |
| RPTP-σ | K67, K68, K70, K71, R76, R96, R99 | ND | 340 Å2 (12 × 28 Å) | Loop, loop, helix | 0.3 nM |
| Slit-2 | R461, R462, K466, R467, K472, K475 | ND | 360 Å2 (15 × 24 Å) | Helix, loop, helix | 80–100M |
| ROBO1 | K69, R117, K122, K123 | ND | 200 Å2 (12 × 17 Å) | Loop, loop, loop | Not reported |
| Thrombin | R93, R101, R126, R165, R233, K236, K240 | H91 | 300 Å2 (12 × 25 Å) | Loop, helix, helix, helix | 7 μM |
| Antithrombin | R46, R47, K114, K125, R129 | N45 | 300 Å(15 × 20 Å) | Loop, loop, helix | 3 nM |
| Protein C inhibitor | R229, K266, R269, K270, K273 | ND | 350 Å(14 × 25 Å) | Loop, helix | Not reported |
| Amyloid precursor protein 1 | K314, R369, K422, R429 | H307, H376, H426, H430, H433 | 420 Å, dimer (13 × 32 Å) | Helix, helix, helix | 800 nM |
| Thrombospondin-1 | R29, K32, R42, R77, K80, K81 | None | 650 Å, dimer (13 × 50 Å) | Strand, loop, strand, strand, loop | 100 nM |
| Annexin A2 | K280, K323 | H93, calcium | 230 Å, (11 × 21 Å) | Helix, helix, loop | 17 nM |
The polar residues that are involved in heparan sulfate (HS) binding were usually identified only in cocrystallization studies because these residues are rarely targeted for site-directed mutagenesis.
Dimensions are approximate. For oligomeric HS-binding proteins (HSBPs), dimensions are for each monomer, and the final area is the sum of the areas of all monomers. In exceptional cases (RAGE, thrombospondin-1, and amyloid), where a continuous HS-binding site (HBS) is formed across the dimer interface, the dimensions are for the composite HBS instead of for individual monomers.
Secondary elements are in the same sequence as the occurrence of the HS-binding residues.
For the sake of consistency, these affinity data are taken from HSBP–heparin binding studies in which heparin was immobilized, whenever available. Abbreviations: FGFR, fibroblast growth factor (FGF) receptor; HGF/SF, hepatocyte growth factor/scatter factor; HMGB, high-mobility group protein B; IL, interleukin; Kd, rate dissociation constant; MCP, monocyte chemoattractant protein; ND, no data; PF4, platelet factor 4; RAGE, receptor for advanced glycation end products; RPTP, receptor protein tyrosine phosphatase; SDF, stromal cell–derived factor; VEGF, vascular endothelial growth factor.
How many lysines and arginines are usually involved?
Electrostatic interactions dominate the interaction of most HSBPs with heparin/HS. The vast majority of characterized HS-binding sites (20 out of 22) contain four to seven lysine or arginine residues. Interestingly, the number of basic residues does not correlate with the affinity of HSBPs for heparin, which also includes the contribution of hydrogen-bonding, van der Waal, and hydrophobic interactions. For example, the HS-binding sites of FGFR1, thrombin, RAGE, and RPTP-σ contain seven basic residues, yet their affinities for heparin range from 0.3 nM to 7 μM (54, 78, 87, 96). In contrast, HS-binding sites containing only four basic residues can have high binding affinity, such as FGF2 and VEGF (121, 133). Note that many HSBPs bind HS as oligomers (more than 50%), which increases valency and the number of basic residues involved in binding. For example, each monomer of PF4 contains six basic residues that interact with HS; thus, a PF4 tetramer docks with HS by way of 24 basic residues (71).
Annexins, which play a role in the fibrinolytic pathway, represent a notable exception to the general rule of four to seven basic residues per HS-binding site. These proteins bind HS in a calcium-dependent manner. The cocrystal structures of annexin A2 and heparin-derived oligosaccharides (up to an octasaccharide) suggest that only two basic residues participate in HS binding (134). Two calcium ions occupy two negatively charged pockets in close proximity to the two basic residues and interact either directly or indirectly with the sulfate and carboxylate groups of the oligosaccharide. Annexin A5, another member of the annexin family, similarly interacts with HS (135). Other examples of calcium-dependent HSBPs include L-selectin, P-selectin, and type V collagen (136–138). However, a direct interaction between calcium and HS has not yet been demonstrated in complexes of these proteins with heparin.
Other polar residues form hydrogen bonds.
Amino acids other than lysine and arginine often contribute to and, in some cases, may be critical for binding HS. Histidine is not commonly found in HS-binding sites (Table 2) but has been demonstrated by mutagenesis in the HS-binding sites in MCP-1 and APLP-1 (59, 139) and in the crystal structures of thrombin and annexin A2 (87, 134). The HS-binding site of APLP-1 is exceptional because it contains five histidines (59). All five residues create either direct or water-mediated hydrogen bonds with sulfate groups (65). Consistent with the crystallization data, mutation of each of the five histidines significantly reduced the salt concentration required for elution from heparin/Sepharose beads, suggesting that the histidines substantially contribute to the binding energy (59).
Polar residues, especially asparagine and glutamine, often make hydrogen bonds with sulfate groups—for example, in FGF1 and FGF2. In both cases, three polar residues participate in hydrogen bonding with sulfate groups (49, 121). A thermodynamic study of HS–FGF2 interaction demonstrated that ionic interactions contribute only 30% of the binding free energy and that hydrogen bonding contributes much of the remaining binding energy (29). The contribution of asparagine and glutamine residues to binding is probably underestimated in most HS-binding sites because these residues are rarely targeted for study. One way to identify potential polar residues in HS-binding sites would be to first define the boundary of HS-binding sites by mutation of lysine and arginine residues, and then to target conserved polar residues within this region by mutagenesis.
What is the size of a heparan sulfate–binding site?
At first glance, it may appear that the size of HS-binding sites varies considerably, given that the buried surface areas range from 200 to 1,050 Å2 (Table 2). However, in many cases, these areas reflect the sum of several individual HS-binding sites (referred to as HS-binding units below) that are present in the oligomeric form of the HSBPs. The vast majority (19 out of 22) of monomeric HS-binding sites have a surface area between 200 to 360 Å2. In some proteins, such as FGF1, HGF, and AT, only one HS-binding unit provides high-affinity binding to HS (49, 103, 132). In contrast, other proteins, especially chemokines and cytokines, use more than one HS-binding unit to assemble a composite HS-binding site. These HSBPs usually interact with HS in their oligomeric form (69).
The size of the HS-binding site does not serve as a good indicator of the binding affinity. High-affinity interactions can be supported by either small HS-binding sites (e.g., FGF2, HGF, RPTP-σ, annexin A2) (76, 78, 121, 134) or large HS-binding sites (e.g., PF4, RAGE) (53, 140). Conversely, some HSBPs exhibiting low affinity for heparin or HS have either small (e.g., thrombin, IL-8) or large (e.g., FGFR1, MCP-1) HS-binding sites (73, 87, 96). It follows that the density of basic residues in the HS-binding site does not necessarily predict affinity as well. For instance, FGFR1 contains seven basic residues in an area of 350 Å2 (per monomer) but has a Kd of only 3 μM (96), whereas RAGE has seven basic residues in an area of 525 Å2 but has a Kd of ~3 nM) (54). The HS-binding sites of thrombin and AT have surface areas of ~300 Å2. Thrombin has a higher density of basic residues, but the affinity of AT for HS is three orders of magnitude greater than the affinity of thrombin for HS (86, 87).
Interestingly, most HS-binding sites have a rectangular or half-cylindrical shape. The short axis is usually between 10 and 16 Å. This axis accommodates HS well because it has a rod-like helical shape with a maximal diameter of 12 Å(Figure 1). Lysine and arginine residues spaced along the binding site typically make salt bridges and/or hydrogen bonds with sulfate groups located on both faces of the heparin helix. In most cases, the long axes of HS-binding sites accord reasonably well with the minimal length of oligosaccharides required for interaction. For example, dp4–dp6 (20–30 Åin length) is the minimal length required for interaction with FGF1, FGF2, and HGF/SF (49, 121, 132). Accordingly, the long axes of the HS-binding sites in these proteins are ~25 Å. The HS-binding site in RAGE has an axis length of 55 Å, which accommodates a dp12 oligosaccharide (54–60 Åin length), the minimal length required for inducing dimerization (53).
Secondary structural elements of heparan sulfate–binding sites.
Generally, HS-binding sites consist of residues contributed by two to four spatially separated structural elements (21 out of 22 proteins listed in Table 2). Loops appear to be the most common secondary structural element, which may reflect their flexibility. In fact, only one HS-binding site (APLP-1) is devoid of a loop (65). α-Helices and β-strands occur less frequently than loops, but in some HS-binding sites they can be the dominant structural elements (e.g., APLP-1, thrombospondin) (65, 75). Specificity for subsequences in HS does not seem to depend on particular secondary structural elements. HSBPs with high selectivity, including FGF2 (binding requires Ido2S), cyclophilin B, and AT (binding requires GlcNS3S), have HS-binding sites that are composed primarily of loops (86, 121, 141).
What is the best way to identify a heparan sulfate–binding site?
Several groups have attempted to find a formula to help identify HS-binding sites in HSBPs (142–145). However, these algorithms suffer from sampling errors introduced by considering only a limited number of HSBPs, from the inclusion of hypothetical HS-binding sites that were not tested experimentally, and from the assumption that a linear sequence of amino acids was responsible for HS binding. An analysis of the experimentally determined HS-binding sites listed in Table 2 makes clear that a single linear sequence does not define a HS-binding site and that two to three primary and secondary elements usually make up the binding site. Thus, topology rather than linear sequence defines most HS-binding sites. Therefore, we suggest that it is best to calculate the surface electrostatic potential by using popular software such as Adaptive Poisson–Boltzmann Solver. In the absence of a three-dimensional structure, homology modeling can be performed with structural threading software.
OPPORTUNITIES AND CHALLENGES
Our knowledge about the physiological functions of HS has exploded during the past 30 years, largely because of the identification and characterization of numerous HSBPs (5). However, among the >300 known HSBPs, only a few have been characterized biochemically (<20%) and even fewer have been characterized structurally (<10%). Several factors holding back the field include a lack of availability of the HSBPs (both in adequate quantities and in their native forms) and a lack of supply of highly purified HS and HS-derived oligosaccharides. We suspect that many other HSBPs remain undiscovered, having escaped detection because of their expression in a tissue-specific, developmentally regulated manner or because of their unique presence in different organisms. Thus, identification and characterization of this large family of proteins remain great challenges.
Characterization of HS–HSBP interactions requires structurally defined oligosaccharides. Most studies have utilized heparin, heparin-derived oligosaccharides, chemically desulfated heparin, HS derived from an arbitrary source, or cell lines or model organisms with defects in specific HS-modifying enzymes. Although these tools afford useful insights into the general structural requirements for binding—for example, a specific class of sulfate groups is important—they provide only a “low-resolution view” of the interaction. Ideally, one would like to obtain atomic resolution by cocrystallization of an HSBP with an appropriately sized HS oligosaccharide. However, most structural studies employ heparin-derived oligosaccharides, in part because heparin is available in large quantities and can be readily cleaved enzymatically or chemically and fractionated by size-exclusion and ion-exchange chromatography. Implicit in this approach are the assumptions that the arrangement of sulfated sugars in HS is represented in heparin, or at least present within the highly sulfated domains buried among the additional sulfate groups, and that groups that are not directly involved in binding will not interfere with the interaction (111). Obviously, these assumptions may not be correct. HS has a heterogeneous domain structure consisting of partially sulfated NS domains separated by NAc domains of variable size and transitional zones, which may not be present in heparin. The presence of peripheral sulfate groups and incorrect orientation of the uronic acids could sterically interfere with binding. Furthermore, it is very difficult to obtain a pure isomer of a heparin or HS-derived oligosaccharide of the correct size (generally dp8–dp12). Isomeric heterogeneity in the oligosaccharide results in heterogeneous crystals or poor resolution of the oligosaccharides in the cocrystals. Investigators have recently made substantial progress in the chemical and chemoenzymatic synthesis of structure-defined HS oligosaccharides (125, 146, 147). Thus, in the near future these methods may give rise to tailor-made oligosaccharides to better characterize the HSBPs.
This review focuses on the interactions of heparin-derived oligosaccharides and HS with HSBPs. However, the more relevant ligands in vivo may consist of cell-surface and extracellular matrix proteoglycans that bear HS chains. Fewer than 20 HSPGs are known (see the side-bar). Given the large number of HSBPs and the presence of multiple chains on most proteoglycans, each HSPG may interact with multiple HSBPs. In some cases, genetic studies have established the relevance of a specific HSPG in a given biological context, but generally we know little about the complexes that exist in vivo (5). Finally, we should keep in mind that most investigations focus on internal segments of the chain as potential binding sites, but in fact the ends of the chains may be essential for binding (e.g., in FGF1/FGFR1 complexes). All cells express heparanase, an enzyme that cleaves HS at specific sites, thereby generating reducing and nonreducing ends derived from the sulfated domains (148). These ends may be present in liberated oligosaccharides or may remain attached to proteoglycan core proteins, thereby serving as the natural carbohydrate ligands for HSBPs.
The biomedical importance of HS–HSBP interactions should not be underestimated. Numerous diseases correlate with changes in HS expression and composition, and there are many examples of inborn errors of metabolism that alter GAG assembly and turnover, causing developmental abnormalities and pathophysiology (5). The biological consequences of altered HS undoubtedly result from alterations in HS–HSBP interactions. Understanding the structure of HS-binding sites in HSBPs will also provide targets for potential inhibitors. One can easily imagine screening of small-molecule libraries for compounds that antagonize HS–HSBP interaction (149). Other classes of inhibitors include tailor-made synthetic HS oligosaccharides, HS mimetics, and monoclonal antibodies directed to HS-binding sites. The development of these agents could prove useful for therapeutic intervention to prevent unwanted activation of an HSBP and as reagents for further exploring the structure and function of HS–HSBP interactions.
HEPARAN SULFATE PROTEOGLYCAN.
HS proteoglycans (HSPGs) consist of a core protein and one or more covalently linked HS chains. Of the 17 HSPGs that have been identified so far, many are membrane associated through either a transmembrane domain (syndecans 1–4, CD44v3, neuropilin, betaglycan) or a GPI anchor (glypicans 1–6). Several HSPGs, including collagen XVIII, agrin, and perlecan, are secreted into the extracellular matrix. One proteoglycan, serglycin, is located in the cytoplasmic secretory granules of endothelial, endocrine, and hematopoietic cells. Whether the identity of the core protein affects the structure of HS remains unresolved. The core proteins of HSPGs are not merely carriers of HS; they can also engage directly in protein binding with either extracellular or intracellular proteins.
ACKNOWLEDGMENTS
Our work was supported by grant 13BGIA14150008 from the American Heart Association (to D.X.) and grants HL107150 and GM093131 (to J.D.E.) from the National Institutes of Health. We acknowledge many helpful comments from Ulf Lindahl and John Gallagher and inspiration from H. Edward Conrad.
Glossary
- HS
heparan sulfate
- GAG
glycosaminoglycan
- HSBP
HS-binding protein
- AT
antithrombin
- CS
chondroitin sulfate
- DS
dermatan sulfate
- dp
degree of polymerization
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Guerardel Y, Czeszak X, Sumanovski LT, Karamanos Y, Popescu O, et al. 2004. Molecular fingerprinting of carbohydrate structure phenotypes of three Porifera proteoglycan-like glyconectins. J. Biol. Chem 279:15591–603 [DOI] [PubMed] [Google Scholar]
- 2.Yamada S, Morimoto H, Fujisawa T, Sugahara K. 2007. Glycosaminoglycans in Hydra magnipapillata (Hydrozoa, Cnidaria): demonstration of chondroitin in the developing nematocyst, sting organelle, and structural characterization of glycosaminoglycans. Glycobiology 17:886–94 [DOI] [PubMed] [Google Scholar]
- 3.Lawrence R, Olson SK, Steele RE, Wang L, Warrior R, et al. 2008. Evolutionary differences in glycosaminoglycan fine structure detected by quantitative glycan reductive isotope labeling. J. Biol. Chem 283:33674–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ori A, Wilkinson MC, Fernig DG. 2011. A systems biology approach for the investigation of the heparin/heparan sulfate interactome. J. Biol. Chem 286:19892–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bishop JR, Schuksz M, Esko JD. 2007. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 446:1030–37 [DOI] [PubMed] [Google Scholar]
- 6.Lin X, Wei G, Shi ZZ, Dryer L, Esko JD, et al. 2000. Disruption of gastrulation and heparan sulfate biosynthesis in EXT1-deficient mice. Dev. Biol 224:299–311 [DOI] [PubMed] [Google Scholar]
- 7.Stickens D, Zak BM, Rougier N, Esko JD, Werb Z. 2005. Mice deficient in Ext2 lack heparan sulfate and develop exostoses. Development 132:5055–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maccarana M, Sakura Y, Tawada A, Yoshida K, Lindahl U. 1996. J. Biol. Chem 271:17804–10 [DOI] [PubMed] [Google Scholar]
- 9.Murphy KJ, Merry CL, Lyon M, Thompson JE, Roberts IS, Gallagher JT. 2004. J. Biol. Chem 279:27239–45 [DOI] [PubMed] [Google Scholar]
- 10.Esko JD, Selleck SB. 2002. Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu. Rev. Biochem 71:435–71 [DOI] [PubMed] [Google Scholar]
- 11.Lawrence R, Brown JR, Al-Mafraji K, Lamanna WC, Beitel JR, et al. 2012. Disease-specific non-reducing end carbohydrate biomarkers for mucopolysaccharidoses. Nat. Chem. Biol 8:197–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carlsson P, Kjellén L. 2012. Heparin biosynthesis. Handb. Exp. Pharmacol 207:23–41 [DOI] [PubMed] [Google Scholar]
- 13.Kreuger J, Kjellén L. 2012. Heparan sulfate biosynthesis: regulation and variability. J. Histochem. Cytochem 60:898–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Esko JD, Sharon N. 2009. Microbial lectins: hemagglutinins, adhesins, and toxins In Essentials of Glycobiology, ed. Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, et al. , pp. 489–500. Cold Spring Harbor, N.Y.: Cold Spring Harb. Lab. [PubMed] [Google Scholar]
- 15.Esko JD, Linhardt RJ. 2009. Proteins that bind sulfated glyocosaminoglycans In Essentials of Glycobiology, ed. Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, et al. , pp. 501–12. Cold Spring Harbor, N.Y.: Cold Spring Harb. Lab. [PubMed] [Google Scholar]
- 16.Ishihara M, Fedarko NS, Conrad HE. 1986. Transport of heparan sulfate into the nuclei of hepatocytes. J. Biol. Chem 261:13575–80 [PubMed] [Google Scholar]
- 17.Richardson TP, Trinkaus-Randall V, Nugent MA. 2001. Regulation of heparan sulfate proteoglycan nuclear localization by fibronectin. J. Cell Sci 114:1613–23 [DOI] [PubMed] [Google Scholar]
- 18.Chen L, Sanderson RD. 2009. Heparanase regulates levels of syndecan-1 in the nucleus. PLoS ONE 4:e4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zong F, Fthenou E, Wolmer N, Hollósi P, Kovalszky I, et al. 2009. Syndecan-1 and FGF-2, but not FGF receptor-1, share a common transport route and co-localize with heparanase in the nuclei of mesenchymal tumor cells. PLoS ONE 4:e7346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Varki A, Etzler ME, Cummings RD, Esko JD. 2009. Discovery and classification of glycan-binding proteins In Essentials of Glycobiology, ed. Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, et al. , pp. 375–86. Cold Spring Harbor, N.Y.: Cold Spring Harb. Lab. [PubMed] [Google Scholar]
- 21.Radek KA, Taylor KR, Gallo RL. 2009. FGF-10 and specific structural elements of dermatan sulfate size and sulfation promote maximal keratinocyte migration and cellular proliferation. Wound Repair Regen. 17:118–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taylor KR, Rudisill JA, Gallo RL. 2005. Structural and sequence motifs in dermatan sulfate for promoting fibroblast growth factor 2 (FGF-2) and FGF-7 activity. J. Biol. Chem 280:5300–6 [DOI] [PubMed] [Google Scholar]
- 23.Trowbridge JM, Rudisill JA, Ron D, Gallo RL. 2002. Dermatan sulfate binds and potentiates activity of keratinocyte growth factor (FGF-7). J. Biol. Chem 277:42815–20 [DOI] [PubMed] [Google Scholar]
- 24.Trowbridge JM, Gallo RL. 2002. Dermatan sulfate: new functions from an old glycosaminoglycan. Glycobiology 12:R117–25 [DOI] [PubMed] [Google Scholar]
- 25.Ori A, Wilkinson MC, Fernig DG. 2008. The heparanome and regulation of cell function: structures, functions and challenges. Front. Biosci 13:4309–38 [DOI] [PubMed] [Google Scholar]
- 26.Faller B, Mely Y, Gerard D, Bieth JG. 1992. Heparin-induced conformational change and activation of mucus proteinase inhibitor. Biochemistry 31:8285–90 [DOI] [PubMed] [Google Scholar]
- 27.Olson ST, Halvorson HR, Björk I. 1991. Quantitative characterization of the thrombin–heparin interaction. Discrimination between specific and nonspecific binding models. J. Biol. Chem 266:6342–52 [PubMed] [Google Scholar]
- 28.Friedrich U, Blom AM, Dahlbäck B, Villoutreix BO. 2001. Structural and energetic characteristics of the heparin-binding site in antithrombotic protein C. J. Biol. Chem 276:24122–28 [DOI] [PubMed] [Google Scholar]
- 29.Thompson LD, Pantoliano MW, Springer BA. 1994. Energetic characterization of the basic fibroblast growth factor–heparin interaction: identification of the heparin binding domain. Biochemistry 33:3831–40 [DOI] [PubMed] [Google Scholar]
- 30.Stanford KI, Bishop JR, Foley EM, Gonzales JC, Niesman IR, et al. 2009. Syndecan-1 is the primary heparan sulfate proteoglycan mediating hepatic clearance of triglyceride-rich lipoproteins in mice. J. Clin. Investig 119:3236–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mascotti DP, Lohman TM. 1995. Thermodynamics of charged oligopeptide–heparin interactions. Biochemistry 34:2908–15 [DOI] [PubMed] [Google Scholar]
- 32.Sheinerman FB, Norel R, Honig B. 2000. Electrostatic aspects of protein–protein interactions. Curr. Opin. Struct. Biol 10:153–59 [DOI] [PubMed] [Google Scholar]
- 33.Duchesne L, Octeau V, Bearon RN, Beckett A, Prior IA, et al. 2012. Transport of fibroblast growth factor 2 in the pericellular matrix is controlled by the spatial distribution of its binding sites in heparan sulfate. PLoS Biol. 10:e1001361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sadir R, Imberty A, Baleux F, Lortat-Jacob H. 2004. Heparan sulfate/heparin oligosaccharides protect stromal cell–derived factor 1 (SDF-1)/CXCL12 against proteolysis induced by CD26/dipeptidyl peptidase IV. J. Biol. Chem 279:43854–60 [DOI] [PubMed] [Google Scholar]
- 35.Lortat-Jacob H, Baltzer F, Grimaud JA. 1996. Heparin decreases the blood clearance of interferon-γ and increases its activity by limiting the processing of its carboxyl-terminal sequence. J. Biol. Chem 271:16139–43 [DOI] [PubMed] [Google Scholar]
- 36.Yan D, Lin X. 2009. Shaping morphogen gradients by proteoglycans. Cold Spring Harb. Perspect. Biol 1:a002493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bellaiche Y, The I, Perrimon N. 1998. Tout-velu is a Drosophila homologue of the putative tumour suppressor EXT-1 and is needed for Hh diffusion. Nature 394:85–88 [DOI] [PubMed] [Google Scholar]
- 38.Baeg GH, Lin X, Khare N, Baumgartner S, Perrimon N. 2001. Heparan sulfate proteoglycans are critical for the organization of the extracellular distribution of Wingless. Development 128:87–94 [DOI] [PubMed] [Google Scholar]
- 39.Takei Y, Ozawa Y, Sato M, Watanabe A, Tabata T. 2004. Three Drosophila EXT genes shape morphogen gradients through synthesis of heparan sulfate proteoglycans. Development 131:73–82 [DOI] [PubMed] [Google Scholar]
- 40.Han C, Yan D, Belenkaya TY, Lin X. 2005. Drosophila glypicans Dally and Dally-like shape the extracellular Wingless morphogen gradient in the wing disc. Development 132:667–79 [DOI] [PubMed] [Google Scholar]
- 41.Lei J, Wan FY, Lander AD, Nie Q. 2011. Robustness of signaling gradient in Drosophila wing imaginal disc. Discret. Contin. Dyn. Syst. B 16:835–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carmeliet P, Ng YS, Nuyens D, Theilmeier G, Brusselmans K, et al. 1999. Impaired myocardial angiogenesis and ischemic cardiomyopathy in mice lacking the vascular endothelial growth factor isoforms VEGF164 and VEGF188. Nat. Med 5:495–502 [DOI] [PubMed] [Google Scholar]
- 43.Bulow HE, Hobert O. 2006. The molecular diversity of glycosaminoglycans shapes animal development. Annu. Rev. Cell Dev. Biol 22:375–407 [DOI] [PubMed] [Google Scholar]
- 44.Lortat-Jacob H. 2009. The molecular basis and functional implications of chemokine interactions with heparan sulphate. Curr. Opin. Struct. Biol 19:543–48 [DOI] [PubMed] [Google Scholar]
- 45.Weber M, Sixt M. 2013. Live cell imaging of chemotactic dendritic cell migration in explanted mouse ear preparations. Methods Mol. Biol 1013:215–26 [DOI] [PubMed] [Google Scholar]
- 46.Goodsell DS, Olson AJ. 2000. Structural symmetry and protein function. Annu. Rev. Biophys. Biomol. Struct 29:105–53 [DOI] [PubMed] [Google Scholar]
- 47.Belov AA, Mohammadi M. 2013. Molecular mechanisms of fibroblast growth factor signaling in physiology and pathology. Cold Spring Harb. Perspect. Biol 5:a015958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mohammadi M, Olsen SK, Ibrahimi OA. 2005. Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev. 16:107–37 [DOI] [PubMed] [Google Scholar]
- 49.DiGabriele AD, Lax I, Chen DI, Svahn CM, Jaye M, et al. 1998. Structure of a heparin-linked biologically active dimer of fibroblast growth factor. Nature 393:812–17 [DOI] [PubMed] [Google Scholar]
- 50.Brown A, Robinson CJ, Gallagher JT, Blundell TL. 2013. Cooperative heparin-mediated oligomerization of fibroblast growth factor 1 (FGF1) precedes recruitment of FGFR2 to ternary complexes. Biophys. J 104:1720–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pellegrini L, Burke DF, Von Delft F, Mulloy B, Blundell TL. 2000. Crystal structure of fibroblast growth factor receptor ectodomain bound to ligand and heparin. Nature 407:1029–34 [DOI] [PubMed] [Google Scholar]
- 52.Beenken A, Eliseenkova AV, Ibrahimi OA, Olsen SK, Mohammadi M. 2012. Plasticity in interactions of fibroblast growth factor 1 (FGF1) N terminus with FGF receptors underlies promiscuity of FGF1. J. Biol. Chem 287:3067–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu D, Young JH, Krahn JM, Song D, Corbett KD, et al. 2013. Stable RAGE–heparan sulfate complexes are essential for signal transduction. Am. Chem. Soc. Chem. Biol 8:1611–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu D, Young J, Song D, Esko JD. 2011. Heparan sulfate is essential for high mobility group protein 1 (HMGB1) signaling by the receptor for advanced glycation end products (RAGE). J. Biol. Chem 286:41736–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thinakaran G, Koo EH. 2008. Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem 283:29615–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Herms J, Anliker B, Heber S, Ring S, Fuhrmann M, et al. 2004. Cortical dysplasia resembling human type 2 lissencephaly in mice lacking all three APP family members. EMBO J. 23:4106–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Soba P, Eggert S, Wagner K, Zentgraf H, Siehl K, et al. 2005. Homo- and heterodimerization of APP family members promotes intercellular adhesion. EMBO J. 24:3624–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dyrks T, Weidemann A, Multhaup G, Salbaum JM, Lemaire HG, et al. 1988. Identification, transmembrane orientation and biogenesis of the amyloid A4 precursor of Alzheimer’s disease. EMBO J. 7:949–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xue Y, Lee S, Wang Y, Ha Y. 2011. Crystal structure of the E2 domain of amyloid precursor protein–like protein 1 in complex with sucrose octasulfate. J. Biol. Chem 286:29748–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gralle M, Botelho MG, Wouters FS. 2009. Neuroprotective secreted amyloid precursor protein acts by disrupting amyloid precursor protein dimers. J. Biol. Chem 284:15016–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mok SS, Sberna G, Heffernan D, Cappai R, Galatis D, et al. 1997. Expression and analysis of heparin-binding regions of the amyloid precursor protein of Alzheimer’s disease. FEBS Lett. 415:303–7 [DOI] [PubMed] [Google Scholar]
- 62.Lee S, Xue Y, Hu J, Wang Y, Liu X, et al. 2011. The E2 domains of APP and APLP1 share a conserved mode of dimerization. Biochemistry 50:5453–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rossjohn J, Cappai R, Feil SC, Henry A, McKinstry WJ, et al. 1999. Crystal structure of the N-terminal, growth factor–like domain of Alzheimer amyloid precursor protein. Nat. Struct. Biol 6:327–31 [DOI] [PubMed] [Google Scholar]
- 64.Dahms SO, Hoefgen S, Roeser D, Schlott B, Gührs KH, Than ME. 2010. Structure and biochemical analysis of the heparin-induced E1 dimer of the amyloid precursor protein. Proc. Natl. Acad. Sci. USA 107:5381–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xue Y, Lee S, Ha Y. 2011. Crystal structure of amyloid precursor-like protein 1 and heparin complex suggests a dual role of heparin in E2 dimerization. Proc. Natl. Acad. Sci. USA 108:16229–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Proudfoot AE, Handel TM, Johnson Z, Lau EK, LiWang P, et al. 2003. Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc. Natl. Acad. Sci. USA 100:1885–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Salanga CL, Handel TM. 2011. Chemokine oligomerization and interactions with receptors and glycosaminoglycans: the role of structural dynamics in function. Exp. Cell Res 317:590–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hoogewerf AJ, Kuschert GS, Proudfoot AE, Borlat F, Clark-Lewis I, et al. 1997. Glycosaminoglycans mediate cell surface oligomerization of chemokines. Biochemistry 36:13570–78 [DOI] [PubMed] [Google Scholar]
- 69.Lortat-Jacob H, Grosdidier A, Imberty A. 2002. Structural diversity of heparan sulfate binding domains in chemokines. Proc. Natl. Acad. Sci. USA 99:1229–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kuschert GS, Hoogewerf AJ, Proudfoot AE, Chung CW, Cooke RM, et al. 1998. Identification of a glycosaminoglycan binding surface on human interleukin-8. Biochemistry 37:11193–201 [DOI] [PubMed] [Google Scholar]
- 71.Zhang X, Chen L, Bancroft DP, Lai CK, Maione TE. 1994. Crystal structure of recombinant human platelet factor 4. Biochemistry 33:8361–66 [DOI] [PubMed] [Google Scholar]
- 72.Ziarek JJ, Veldkamp CT, Zhang F, Murray NJ, Kartz GA, et al. 2013. Heparin oligosaccharides inhibit chemokine (CXC motif) ligand 12 (CXCL12) cardioprotection by binding orthogonal to the dimerization interface, promoting oligomerization, and competing with the chemokine (CXC motif) receptor 4 (CXCR4) N terminus. J. Biol. Chem 288:737–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lau EK, Paavola CD, Johnson Z, Gaudry JP, Geretti E, et al. 2004. Identification of the glycosaminoglycan binding site of the CC chemokine, MCP-1: implications for structure and function in vivo. J. Biol. Chem 279:22294–305 [DOI] [PubMed] [Google Scholar]
- 74.Goodger SJ, Robinson CJ, Murphy KJ, Gasiunas N, Harmer NJ, et al. 2008. Evidence that heparin saccharides promote FGF2 mitogenesis through two distinct mechanisms. J. Biol. Chem 283:13001–8 [DOI] [PubMed] [Google Scholar]
- 75.Tan K, Duquette M, Liu JH, Shanmugasundaram K, Joachimiak A, et al. 2008. Heparin-induced cisand trans-dimerization modes of the thrombospondin-1 N-terminal domain. J. Biol. Chem 283:3932–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lietha D, Chirgadze DY, Mulloy B, Blundell TL, Gherardi E. 2001. Crystal structures of NK1–heparin complexes reveal the basis for NK1 activity and enable engineering of potent agonists of the MET receptor. EMBO J. 20:5543–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vander Kooi CW, Jusino MA, Perman B, Neau DB, Bellamy HD, Leahy DJ. 2007. Structural basis for ligand and heparin binding to neuropilin B domains. Proc. Natl. Acad. Sci. USA 104:6152–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Coles CH, Shen Y, Tenney AP, Siebold C, Sutton GC, et al. 2011. Proteoglycan-specific molecular switch for RPTPσ clustering and neuronal extension. Science 332:484–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shaw AS, Filbert EL. 2009. Scaffold proteins and immune-cell signalling. Nat. Rev. Immunol 9:47–56 [DOI] [PubMed] [Google Scholar]
- 80.Jackson CM, Nemerson Y. 1980. Blood coagulation. Annu. Rev. Biochem 49:765–811 [DOI] [PubMed] [Google Scholar]
- 81.Rau JC, Beaulieu LM, Huntington JA, Church FC. 2007. Serpins in thrombosis, hemostasis and fibrinolysis. J. Thromb. Haemost 5(Suppl. 1):102–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Björk I, Lindahl U. 1982. Mechanism of the anticoagulant action of heparin. Mol. Cell Biochem 48:161–82 [DOI] [PubMed] [Google Scholar]
- 83.Olson ST, Björk I. 1991. Predominant contribution of surface approximation to the mechanism of heparin acceleration of the antithrombin–thrombin reaction. Elucidation from salt concentration effects. J. Biol. Chem 266:6353–64 [PubMed] [Google Scholar]
- 84.Li W, Johnson DJ, Esmon CT, Huntington JA. 2004. Structure of the antithrombin–thrombin–heparin ternary complex reveals the antithrombotic mechanism of heparin. Nat. Struct. Mol. Biol 11:857–62 [DOI] [PubMed] [Google Scholar]
- 85.Bray B, Lane DA, Freyssinet JM, Pejler G, Lindahl U. 1989. Anti-thrombin activities of heparin. Effect of saccharide chain length on thrombin inhibition by heparin cofactor II and by antithrombin. Biochem. J 262:225–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Johnson DJ, Li W, Adams TE, Huntington JA. 2006. Antithrombin–S195A factor Xa-heparin structure reveals the allosteric mechanism of antithrombin activation. EMBO J. 25:2029–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Carter WJ, Cama E, Huntington JA. 2005. Crystal structure of thrombin bound to heparin. J. Biol. Chem 280:2745–49 [DOI] [PubMed] [Google Scholar]
- 88.Dementiev A, Petitou M, Herbert JM, Gettins PG. 2004. The ternary complex of antithrombin–anhydrothrombin–heparin reveals the basis of inhibitor specificity. Nat. Struct. Mol. Biol 11:863–67 [DOI] [PubMed] [Google Scholar]
- 89.Neese LL, Wolfe CA, Church FC. 1998. Contribution of basic residues of the D and H helices in heparin binding to protein C inhibitor. Arch. Biochem. Biophys 355:101–8 [DOI] [PubMed] [Google Scholar]
- 90.Li W, Adams TE, Nangalia J, Esmon CT, Huntington JA. 2008. Molecular basis of thrombin recognition by protein C inhibitor revealed by the 1.6-Å structure of the heparin-bridged complex. Proc. Natl. Acad. Sci. USA 105:4661–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yang L, Manithody C, Rezaie AR. 2002. Contribution of basic residues of the 70–80-loop to heparin binding and anticoagulant function of activated protein C. Biochemistry 41:6149–57 [DOI] [PubMed] [Google Scholar]
- 92.Li W, Huntington JA. 2008. The heparin binding site of protein C inhibitor is protease-dependent. J. Biol. Chem 283:36039–45 [DOI] [PubMed] [Google Scholar]
- 93.Ibrahimi OA, Zhang F, Hrstka SC, Mohammadi M, Linhardt RJ. 2004. Kinetic model for FGF, FGFR, and proteoglycan signal transduction complex assembly. Biochemistry 43:4724–30 [DOI] [PubMed] [Google Scholar]
- 94.Kalinina J, Dutta K, Ilghari D, Beenken A, Goetz R, et al. 2012. The alternatively spliced acid box region plays a key role in FGF receptor autoinhibition. Structure 20:77–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Powell AK, Fernig DG, Turnbull JE. 2002. Fibroblast growth factor receptors 1 and 2 interact differently with heparin/heparan sulfate: implications for dynamic assembly of a ternary signaling complex. J. Biol. Chem 277:28554–63 [DOI] [PubMed] [Google Scholar]
- 96.Schlessinger J, Plotnikov AN, Ibrahimi OA, Eliseenkova AV, Yeh BK, et al. 2000. Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol. Cell 6:743–50 [DOI] [PubMed] [Google Scholar]
- 97.Robinson CJ, Harmer NJ, Goodger SJ, Blundell TL, Gallagher JT. 2005. Cooperative dimerization of fibroblast growth factor 1 (FGF1) upon a single heparin saccharide may drive the formation of 2:2:1 FGF1·FGFR2c·heparin ternary complexes. J. Biol. Chem 280:42274–82 [DOI] [PubMed] [Google Scholar]
- 98.Olsen SK, Li JY, Bromleigh C, Eliseenkova AV, Ibrahimi OA, et al. 2006. Structural basis by which alternative splicing modulates the organizer activity of FGF8 in the brain. Genes Dev. 20:185–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fukuhara N, Howitt JA, Hussain SA, Hohenester E. 2008. Structural and functional analysis of slit and heparin binding to immunoglobulin-like domains 1 and 2 of Drosophila Robo. J. Biol. Chem 283:16226–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.del Sol A, Tsai CJ, Ma B, Nussinov R. 2009. The origin of allosteric functional modulation: multiple pre-existing pathways. Structure 17:1042–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tsai CJ, del Sol A, Nussinov R. 2008. Allostery: absence of a change in shape does not imply that allostery is not at play. J. Mol. Biol 378:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Langdown J, Belzar KJ, Savory WJ, Baglin TP, Huntington JA. 2009. The critical role of hinge-region expulsion in the induced-fit heparin binding mechanism of antithrombin. J. Mol. Biol 386:1278–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jin L, Abrahams JP, Skinner R, Petitou M, Pike RN, Carrell RW. 1997. The anticoagulant activation of antithrombin by heparin. Proc. Natl. Acad. Sci. USA 94:14683–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Johnson DJ, Langdown J, Huntington JA. 2010. Molecular basis of factor IXa recognition by heparin-activated antithrombin revealed by a 1.7-Åstructure of the ternary complex. Proc. Natl. Acad. Sci. USA 107:645–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Al-Mohanna F, Parhar R, Kotwal GJ. 2001. Vaccinia virus complement control protein is capable of protecting xenoendothelial cells from antibody binding and killing by human complement and cytotoxic cells. Transplantation 71:796–801 [DOI] [PubMed] [Google Scholar]
- 106.Khan S, Nan R, Gor J, Mulloy B, Perkins SJ. 2012. Bivalent and co-operative binding of complement factor H to heparan sulfate and heparin. Biochem. J 444:417–28 [DOI] [PubMed] [Google Scholar]
- 107.Ganesh VK, Smith SA, Kotwal GJ, Murthy KH. 2004. Structure of Vaccinia complement protein in complex with heparin and potential implications for complement regulation. Proc. Natl. Acad. Sci. USA 101:8924–29 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 108.Murthy KHM, Smith SA, Ganesh VK, Judge KW, Mullin N, et al. 2001. Crystal structure of a complement control protein that regulates both pathways of complement activation and binds heparan sulfate proteoglycans. Cell 104:301–11 [DOI] [PubMed] [Google Scholar]
- 109.Meri S, Pangburn MK. 1990. Discrimination between activators and nonactivators of the alternative pathway of complement: regulation via a sialic acid/polyanion binding site on factor H. Proc. Natl. Acad. Sci. USA 87:3982–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kreuger J, Spillmann D, Li JP, Lindahl U. 2006. Interactions between heparan sulfate and proteins: the concept of specificity. J. Cell Biol 174:323–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lindahl U, Li JP. 2009. Interactions between heparan sulfate and proteins—design and functional implications. Int. Rev. Cell Mol. Biol 276:105–59 [DOI] [PubMed] [Google Scholar]
- 112.Richard B, Swanson R, Olson ST. 2009. The signature 3-O-sulfo group of the anticoagulant heparin sequence is critical for heparin binding to antithrombin but is not required for allosteric activation. J. Biol. Chem 284:27054–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Turnbull JE, Fernig DG, Ke Y, Wilkinson MC, Gallagher JT. 1992. Identification of the basic fibroblast growth factor binding sequence in fibroblast heparan sulfate. J. Biol. Chem 267:10337–41 [PubMed] [Google Scholar]
- 114.Lortat-Jacob H, Turnbull JE, Grimaud JA. 1995. Molecular organization of the interferon γ–binding domain in heparan sulphate. Biochem. J 310:497–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Spillmann D, Witt D, Lindahl U. 1998. Defining the interleukin-8-binding domain of heparan sulfate. J. Biol. Chem 273:15487–93 [DOI] [PubMed] [Google Scholar]
- 116.Kamimura K, Koyama T, Habuchi H, Ueda R, Masu M, et al. 2006. Specific and flexible roles of heparan sulfate modifications in Drosophila FGF signaling. J. Cell Biol 174:773–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Liu J, Shworak NW, Sinay P, Schwartz JJ, Zhang L, et al. 1999. Expression of heparan sulfate D-glucosaminyl 3-O-sulfotransferase isoforms reveals novel substrate specificities. J. Biol. Chem 274:5185–92 [DOI] [PubMed] [Google Scholar]
- 119.Neugebauer JM, Cadwallader AB, Amack JD, Bisgrove BW, Yost HJ. 2013. Differential roles for 3-OSTs in the regulation of cilia length and motility. Development 140:3892–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jemth P, Kreuger J, Kusche-Gullberg M, Sturiale L, Giménez-Gallego G, Lindahl U. 2002. Biosynthetic oligosaccharide libraries for identification of protein-binding heparan sulfate motifs—exploring the structural diversity by screening for fibroblast growth factor (FGF) 1 and FGF2 binding. J. Biol. Chem 277:30567–73 [DOI] [PubMed] [Google Scholar]
- 121.Faham S, Hileman RE, Fromm JR, Linhardt RJ, Rees DC. 1996. Heparin structure and interactions with basic fibroblast growth factor. Science 271:1116–20 [DOI] [PubMed] [Google Scholar]
- 122.Pye DA, Vives RR, Turnbull JE, Hyde P, Gallagher JT. 1998. J. Biol. Chem 273:22936–42 [DOI] [PubMed] [Google Scholar]
- 123.Jastrebova N, Vanwildemeersch M, Lindahl U, Spillmann D. 2010. J. Biol. Chem 285:26842–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Luo Y, Ye S, Kan M, McKeehan WL. 2006. J. Cell. Biochem 97:1241–58 [DOI] [PubMed] [Google Scholar]
- 125.Dulaney SB, Huang X. 2012. Strategies in synthesis of heparin/heparan sulfate oligosaccharides: 2000–present. Adv. Carbohydr. Chem. Biochem 67:95–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Turnbull JE, Gallagher JT. 1991. Sequence analysis of heparan sulphate indicates defined location of N-sulphated glucosamine and iduronate 2-sulphate residues proximal to the protein-linkage region. Biochem. J 277:297–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Stringer SE, Gallagher JT. 1997. Specific binding of the chemokine platelet factor 4 to heparan sulfate. J. Biol. Chem 272:20508–14 [DOI] [PubMed] [Google Scholar]
- 128.Stringer SE, Forster MJ, Mulloy B, Bishop CR, Graham GJ, Gallagher JT. 2002. Characterization of the binding site on heparan sulfate for macrophage inflammatory protein 1α. Blood 100:1543–50 [PubMed] [Google Scholar]
- 129.Axelsson J, Xu D, Na Kang B, Nussbacher JK, Handel TM, et al. 2012. Inactivation of heparan sulfate 2-O-sulfotransferase accentuates neutrophil infiltration during acute inflammation in mice. Blood 120:1742–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Carter NM, Ali S, Kirby JA. 2003. Endothelial inflammation: the role of differential expression of N-deacetylase/N-sulphotransferase enzymes in alteration of the immunological properties of heparan sulphate. J. Cell Sci 116:3591–600 [DOI] [PubMed] [Google Scholar]
- 131.Krenn EC, Wille I, Gesslbauer B, Poteser M, van Kuppevelt TH, Kungl AJ. 2008. Glycanogenomics: a qPCR-approach to investigate biological glycan function. Biochem. Biophys. Res. Commun 375:297–302 [DOI] [PubMed] [Google Scholar]
- 132.Catlow KR, Deakin JA, Wei Z, Delehedde M, Fernig DG, et al. 2008. Interactions of hepatocyte growth factor/scatter factor with various glycosaminoglycans reveal an important interplay between the presence of iduronate and sulfate density. J. Biol. Chem 283:5235–48 [DOI] [PubMed] [Google Scholar]
- 133.Krilleke D, Ng YS, Shima DT. 2009. The heparin-binding domain confers diverse functions of VEGF-A in development and disease: a structure–function study. Biochem. Soc. Trans 37:1201–6 [DOI] [PubMed] [Google Scholar]
- 134.Shao C, Zhang F, Kemp MM, Linhardt RJ, Waisman DM, et al. 2006. Crystallographic analysis of calcium-dependent heparin binding to annexin A2. J. Biol. Chem 281:31689–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Capila I, VanderNoot VA, Mealy TR, Seaton BA, Linhardt RJ. 1999. Interaction of heparin with annexin V. FEBS Lett. 446:327–30 [DOI] [PubMed] [Google Scholar]
- 136.Norgard-Sumnicht KE, Varki NM, Varki A. 1993. Calcium-dependent heparin-like ligands for L-selectin in nonlymphoid endothelial cells. Science 261:480–83 [DOI] [PubMed] [Google Scholar]
- 137.Koenig A, Norgard-Sumnicht K, Linhardt R, Varki A. 1998. Differential interactions of heparin and heparan sulfate glycosaminoglycans with the selectins. Implications for the use of unfractionated and low molecular weight heparins as therapeutic agents. J. Clin. Investig 101:877–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ricard-Blum S, Beraud M, Raynal N, Farndale RW, Ruggiero F. 2006. Structural requirements for heparin/heparan sulfate binding to type V collagen. J. Biol. Chem 281:25195–204 [DOI] [PubMed] [Google Scholar]
- 139.Chakravarty L, Rogers L, Quach T, Breckenridge S, Kolattukudy PE. 1998. Lysine 58 and histidine 66 at the C-terminal α-helix of monocyte chemoattractant protein 1 are essential for glycosaminoglycan binding. J. Biol. Chem 273:29641–47 [DOI] [PubMed] [Google Scholar]
- 140.Sheng GJ, Oh YI, Chang SK, Hsieh-Wilson LC. 2013. Tunable heparan sulfate mimetics for modulating chemokine activity. J. Am. Chem. Soc 135:10898–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hanoulle X, Melchior A, Sibille N, Parent B, Denys A, et al. 2007. Structural and functional characterization of the interaction between cyclophilin B and a heparin-derived oligosaccharide. J. Biol. Chem 282:34148–58 [DOI] [PubMed] [Google Scholar]
- 142.Cardin AD, Weintraub HJ. 1989. Molecular modeling of protein–glycosaminoglycan interactions. Arteriosclerosis 9:21–32 [DOI] [PubMed] [Google Scholar]
- 143.Sobel M, Soler DF, Kermode JC, Harris RB. 1992. Localization and characterization of a heparin binding domain peptide of human von Willebrand factor. J. Biol. Chem 267:8857–62 [PubMed] [Google Scholar]
- 144.Margalit H, Fischer N, Ben-Sasson SA. 1993. Comparative analysis of structurally defined heparin binding sequences reveals a distinct spatial distribution of basic residues. J. Biol. Chem 268:19228–31 [PubMed] [Google Scholar]
- 145.Munoz EM, Linhardt RJ. 2004. Heparin-binding domains in vascular biology. Arterioscler. Thromb. Vasc. Biol 24:1549–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Moon AF, Xu Y, Woody SM, Krahn JM, Linhardt RJ, et al. 2012. Dissecting the substrate recognition of 3-O-sulfotransferase for the biosynthesis of anticoagulant heparin. Proc. Natl. Acad. Sci. USA 109:5265–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Xu Y, Masuko S, Takieddin M, Xu H, Liu R, et al. 2011. Chemoenzymatic synthesis of homogeneous ultralow molecular weight heparins. Science 334:498–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Peterson SB, Liu J. 2013. Multi-faceted substrate specificity of heparanase. Matrix Biol. 32:223–27 [DOI] [PubMed] [Google Scholar]
- 149.Schuksz M, Fuster MM, Brown JR, Crawford BE, Ditto DP, et al. 2008. Surfen, a small molecule antagonist of heparan sulfate. Proc. Natl. Acad. Sci. USA 105:13075–80 [DOI] [PMC free article] [PubMed] [Google Scholar]