

Abstract
The global pandemic of novel coronavirus disease 2019 (COVID‐19) caused by severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) has created an urgent need for effective antivirals. Remdesivir (formerly GS‐5734) is a nucleoside analogue pro‐drug currently being evaluated in COVID‐19 clinical trials. Its unique structural features allow high concentrations of the active triphosphate metabolite to be delivered intracellularly and it evades proofreading to successfully inhibit viral RNA synthesis. In pre‐clinical models, remdesivir has demonstrated potent antiviral activity against diverse human and zoonotic β‐coronaviruses, including SARS‐CoV‐2. In this article, we critically review available data on remdesivir with an emphasis on biochemistry, pharmacology, pharmacokinetics, and in vitro activity against coronaviruses as well as clinical experience and current progress in COVID‐19 clinical trials.
Keywords: Remdesivir, GS‐5734, COVID‐19, SARS‐CoV‐2, coronavirus, severe acute respiratory syndrome
The global pandemic of novel coronavirus disease 2019 (COVID‐19) caused by severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) has created an urgent need for effective antivirals. 1 , 2 A wide variety of novel and repurposed agents are currently being evaluated in clinical trials and anecdotal reports of off‐label and compassionate use of a number of these agents have emerged. 3 , 4 Remdesivir (formerly GS‐5734) is a monophosphoramidate nucleoside analogue pro‐drug that was originally developed in response to the 2014–2016 Ebola outbreak in West Africa. 5 , 6 It has shown broad‐spectrum activity against human and zoonotic coronaviruses in pre‐clinical models and has been prioritized for inclusion in COVID‐19 clinical trials. 4 , 6 , 7 , 8 , 9 The purpose of this article is to critically review available data on remdesivir with an emphasis on biochemistry, pharmacology, pharmacokinetics (PK), and in vitro activity against coronaviruses as well as clinical experience and current progress in COVID‐19 clinical trials.
1. Data Sources
A literature search of PubMed was conducted on April 24, 2020, and updated on May 26, 2020, using the search terms “remdesivir,” “GS‐5734,” and “GS‐441524.” Results were limited to articles available in English. The reference lists of relevant articles were also examined to identify sources not captured in the literature search. Additional data were obtained from ClinicalTrials.gov, bioRxiv, the World Health Organization (WHO), the European Medicines Agency (EMA), and the US Food and Drug Administration (FDA).
2. Chemistry and Pharmacology
Remdesivir (formerly GS‐5734, Figure 1) is a phosphoramidate pro‐drug of a 1′‐cyano‐substituted nucleoside analogue (GS‐441524). It inhibits viral replication by competing with endogenous nucleotides for incorporation into replicating viral RNA via RNA dependent RNA polymerase (RdRp). 6 The RdRp non‐structural protein (nsp12) is highly conserved across coronaviruses making it an attractive target for broad‐spectrum antiviral drugs. Once inside cells, remdesivir undergoes rapid metabolic conversion by intracellular kinases to its active nucleoside triphosphate metabolite (GS‐443902). In general, the rate‐limiting step for activation of nucleoside analogues is the generation of the nucleoside monophosphate. 6 Nucleoside phosphoramidates, like remdesivir (and GS‐441524), are bioisosteres of monophosphates and are thereby able to bypass this rate‐limiting step. 6 Nucleoside phosphoramidates, however, must be administered as pro‐drugs to mask the charged phosphonate group and allow faster cell entry. In the case of remdesivir, the negative charge is masked by the 2‐ethylbutyl and L‐alanine groups, which are rapidly removed by intracellular esterases. In addition, the 1′‐CN group on remdesivir and its metabolites confers high selectivity for RdRp compared to human polymerases (Figure 1). 6
Figure 1.
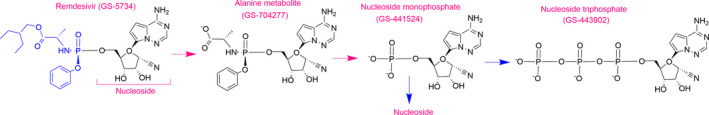
Chemical structures of remdesivir and its metabolites.
The development of effective nucleoside analogues against coronaviruses has been particularly challenging due to the presence of a unique exoribonuclease (ExoN). ExoN functions as a proofreading enzyme correcting errors in the growing RNA chain. 10 , 11 The poor in vitro activity of ribavirin against coronaviruses, for example, has been attributed to its removal by ExoN. 11 Remdesivir is more active against viruses lacking ExoN but is able to partly evade proofreading and maintain potent antiviral activity in the presence of ExoN. This was nicely illustrated in a β‐coronavirus murine hepatitis virus (MHV) model. 11 Compared to wild‐type MHV (EC50 0.087 μmol/L), viruses lacking ExoN were approximately 4‐fold more sensitive to remdesivir inhibition (EC50 0.019 μmol/L). The reason remdesivir’s activity is only modestly decreased by ExoN relates to two unique properties. First, remdesivir is incorporated into replicating RNA more efficiently than natural nucleotides. 10 , 12 , 13 This was shown in a series of kinetic studies that determined the selectivity of β‐coronaviruses for remdesivir vs. natural nucleotides. 10 , 12 In this context selectivity was defined as the ratio of the parameters Vmax (reflecting the maximal velocity of nucleotide incorporation) to Km (reflecting the substrate concentration at which the velocity of incorporation is half Vmax) for a single incorporation of a natural nucleotide over a nucleotide analogue. Selectivity values below 1 indicate the analogue is incorporated more efficiently by RdRp than the natural nucleotide while values above 1 suggest the opposite. Selectivity values were low for SARS‐CoV‐1, SARS‐CoV‐2, and Middle Eastern respiratory syndrome coronavirus (MERS‐CoV) (0.32, 0.26, and 0.35, respectively). Interestingly, selectivity values were higher for Ebola virus (4) and for other nucleotide analogues with SARS‐CoV‐2 (favipiravir = 570 and ribavirin >> 1000). 10
The second reason that remdesivir is able to partly evade ExoN is because it functions as a non‐obligate or delayed RNA chain terminator. 10 , 12 , 13 Delayed chain termination occurs when a nucleotide analogue has a free 3′‐OH group required for the addition of natural nucleotides. The incorporation of the delayed chain terminator, however, perturbs the RNA structure, and synthesis is halted at some point downstream. 13 In SARS‐CoV‐1, SARS‐CoV‐2, and MERS‐CoV, remdesivir (GS‐443902) incorporation consistently results in chain termination after three additional nucleotides are added. 10 , 12 It is thought that these nucleotides provide protection from ExoN excision. 10 , 12
3. Pharmacokinetics
As described in the chemistry and pharmacology section, remdesivir is a pro‐drug; concentrations decline rapidly after IV administration (plasma half‐life, T½ ~1 hr), followed by the sequential appearance of the intermediate alanine metabolite GS‐704277 and the nucleoside monophosphate metabolite GS‐441524 (plasma T½ 24.5 hrs) (Figure 1). Inside cells, GS‐441524 is rapidly converted to the pharmacologically active triphosphate analogue, GS‐443902, which has a prolonged intracellular T ½ (peripheral blood mononuclear cell, PBMC T½ ~ 40 hrs). Both remdesivir and GS‐441524 exhibit linear PK following single doses between 3 mg and 225 mg and no remdesivir accumulation was observed following once daily dosing for up to 5 days. By contrast, GS‐441524 reaches steady state around day 4 and accumulates by ~2‐fold after multiple once daily dosing. 3
The remdesivir dosing regimen being evaluated in clinical trials (200 mg IV on day 1, then 100 mg IV on days 2 through 5 or 10) was substantiated by in vitro data and bridging the PK with the rhesus monkey experience to humans. 7 , 8 , 14 Table 1 summarizes pertinent PK parameters of remdesivir and metabolite GS‐441524, which were derived from single‐ and multiple‐dose studies in healthy human adult volunteers. 14 As shown, remdesivir Cmax values are many fold above concentrations required in vitro to inhibit SARS‐CoV‐2 replication by 50% and 90% (EC50 0.137–0.77 μmol/L, EC90 1.76 μmol/L, see microbiology section). 14 , 15
Table 1.
Plasma Pharmacokinetic Parameters of Remdesivir and GS‐441524 Following Remdesivir 200 mg IV over 30 min on day 1, then 100 mg IV daily over 30 min on days 2 to 5 in Healthy Human Subjects (adapted from 3 )
| Parameter | Remdesivir | GS‐441524 | ||
|---|---|---|---|---|
| Day 1 | Day 5 | Day 1 | Day 5 | |
| Cmax |
5.44 µg/mL 9.03 µM b |
2.61 µg/mL 4.33 µM b |
0.15 µg/mL 0.52 µM b |
0.14 µg/mL 0.48 µM b |
| AUC a |
2.92 h* µg/mL 4.85 µM b |
1.56 h* µg/mL 2.59 µM b |
2.24 h* µg/mL 7.69 µM b |
2.23 h* µg/mL 7.66 µM b |
| T ½ | 0.98 (0.82–1.03) c h | 0.89 (0.82–1.09) c h | N/A | 25.30 (24.10–30.32) c h |
| Free fraction | 12.1% | 85–127% | ||
AUC = area under the concentration time curve; T½ = half‐life.
AUC24 presented on day 1, AUC tau presented on day 5.
Molar concentrations calculated using molecular weights of 602.6 g/mol and 291.26 g/mol for remdesivir and GS‐441524, respectively.
Interquartile range.
Due to the near complete first‐pass effect of phosphoramidates, remdesivir is expected to have poor oral bioavailability. 6 Plasma protein binding for remdesivir is moderate (free fraction of 12.1%). By contrast, metabolites GS‐704277 and GS‐441524 exhibit low plasma protein binding with mean free fractions ≥ 85%. 14 In cynomolgus monkeys, radiolabeled remdesivir or its metabolites were detectable in the testes, epididymis, eyes, and brain 4 hours after a 10‐mg/kg dose (equivalent to 200 mg in humans). 16 Levels in the brain were significantly lower than in other tissues but accumulated over time. 16 Distribution studies in humans have not yet been reported.
Remdesivir is a substrate of several cytochrome P450 enzymes in vitro (see drug interactions section), however clinical implications are unclear since the pro‐drug is rapidly metabolized by plasma hydrolases. 14 By similar reasoning, the effect of hepatic impairment on remdesivir plasma levels should be low although specific studies have not been conducted in patients with hepatic impairment and the drug is contraindicated in patients with severe hepatic impairment. 14 Metabolism of metabolites GS‐704277, GS‐441524, and GS‐443902 has not been characterized.
Remdesivir exhibits low renal excretion (< 10%). However, 49% of a radiolabeled dose was recovered as GS‐441524 in urine. 14 Theoretically, plasma exposure of GS‐441524 may be increased in patients with renal impairment. Remdesivir formulations contain sulfobutylether β‐cyclodextrin sodium (SBECD) as a solubility enhancer. 14 Formulations containing SBECD have historically been cautioned against in patients with renal impairment, although clinical data suggest SBECD accumulation does not increase the risk of acute kidney injury. 17 There are no recommendations for dose adjustments in patients with mild to moderate renal impairment at this time. Under the FDA emergency use authorization guidance, remdesivir is not recommended in patients with an estimated glomerular filtration rate (eGFR) < 30 ml/min or serum creatinine ≥ 1 mg/dl unless the potential benefit outweighs the potential risk. 18 Patients with an eGFR < 30 ml/min and those who are receiving hemodialysis or hemofiltration are excluded from the EMA compassionate use program. 14 There are no PK data available for children or women who are pregnant or breast feeding.
4. Microbiology
Remdesivir has demonstrated broad‐spectrum activity against a diverse panel of zoonotic and clinically relevant human coronaviruses including SARS‐CoV‐1, SARS‐CoV‐2, and MERS‐CoV with micromolar EC50 or IC50 values in multiple in vitro systems. 3 , 9 , 11 , 19 , 20 In human airway epithelial cell cultures, for example, remdesivir inhibited SARS‐CoV‐1 and MERS‐CoV replication with IC50 values of 0.069 and 0.074 μmol/L, respectively. 9 Emerging data suggest remdesivir also exhibits potent activity against SARS‐CoV‐2. 14 , 15 , 21 In testing performed by the Chinese CDC in collaboration with the manufacturer, Gilead Sciences, Inc., using Vero E6 cells (a cell line from green monkey kidney epithelial cells that is commonly used to evaluate antiviral activity), the EC50 value was 0.137 μmol/L against SARS‐CoV‐2. 14 Researchers recently evaluated the impact remdesivir and six other compounds on SARS‐CoV‐2 viral titers and cytotoxicity and infection rates. 15 Remdesivir demonstrated the most potent activity with EC50 and EC90 values of 0.77 and 1.76 μmol/L, respectively. 15 A second study assessed the antiviral activity of remdesivir and 15 other candidate compounds against SARS‐CoV‐2. Whereas earlier remdesivir studies have fitted viral load in linear scale and quantified the percentage of viral replication inhibition under increasing drug concentrations, 14 , 15 in this study viral load was fitted in logarithm scales (log10TCID50/ml and log10 viral RNA copies/ml). Using this scale, EC50 values were many fold higher at 23.15 and 26.90 μmol/L, respectively. 21 It is uncertain which method is most reflective of activity in vivo. It should also be remembered that EC50 and IC50 values represent a single point on the dose‐response curve and the slope of the curve may be more important when evaluating potency. 22 This relationship is generally neglected when assessing in vitro antiviral activity. Furthermore, clinical outcomes may depend on whether > 90% inhibition is achieved but EC90 values are reported in a minority of studies. It is reassuring that the EC90 value reported (1.76 μmol/L) is achievable with the dosing regimen under evaluation (see PK section). 14 , 15
5. Resistance
The development of remdesivir resistance in coronaviruses has been assessed by cell culture in MHV, which has similar EC50 values to SARS‐CoV‐1, SARS‐CoV‐2, and MERS‐CoV. 11 Following >20 in vitro passages, two nonsynonymous mutations were selected in the nsp12 RdRp: F476L and V553L. Neither of the mutations directly altered RdRp’s catalytic site or substrate binding pocket, but they did cause minor structural alterations that are thought to impact RdRp’s fidelity checking step before catalysis. 11 , 13 Compared to wild‐type virus, these mutations conferred 2.4‐fold and 5‐fold reduced susceptibility to remdesivir, respectively, while the double mutant showed 5.6‐fold reduced susceptibility in vitro. 11 The EC50 values of the mutants (0.057–0.13 μmol/L), however, remained below achievable human drug exposures. 3 , 11 , 13 Furthermore, the mutations appear to confer a fitness cost, with wild‐type virus rapidly outcompeting the mutants in the absence of remdesivir. Of concern, however, the affected residues are conserved across coronaviruses raising the possibility of a common pathway to resistance. In fact, substitutions at homologous SARS‐CoV‐1 residues conferred a 6‐fold decrease in susceptibility to remdesivir (EC50 0.01 μmol/L → 0.06 μmol/L). 11 No data specific to SARS‐CoV‐2 remdesivir resistance have been published at the time of writing.
6. Animal Studies
Remdesivir’s efficacy against respiratory diseases caused by β‐coronaviruses has been evaluated in both rodent and rhesus monkey models. 7 , 8 , 9 , 14 , 19
Before summarizing these studies, the reader should be aware that rodent models are not ideal for evaluating remdesivir efficacy against β‐coronaviruses. First, high levels of serum esterases rapidly degrade the pro‐drug (T½ ~ 5 min). 9 , 16 Investigators have attempted to overcome this by using esterase knock‐out mice. 9 This improves plasma stability (T½ ~25 min), but tissue levels of active metabolites are still ~10‐fold lower. 9 Second, the active triphosphate metabolite (GS‐443902) has a significantly shorter T½ in the mouse lung (3 hrs) compared to human lung cells and non‐human primate lungs (T½ 20–40 hrs). 9 , 14 Finally, mice are naturally resistant to infection by MERS‐CoV due to the inability of the spike protein binding domain to interact with the mouse DPP4 receptor. Therefore, transgenic mice harboring a modified humanized DPP4 must be used. 19
These limitations notwithstanding, researchers evaluated prophylactic and therapeutic remdesivir against SARS‐CoV‐1 in an esterase‐deficient mouse model. 9 Compared to control mice, prophylactic administration of remdesivir (24 hrs before viral inoculation) resulted in less weight loss, lower viral lung titers, and reduced SARS‐CoV‐1–induced lung pathology. 9 Similarly, therapeutic remdesivir (administered 1 day post‐inoculation) significantly diminished weight loss and viral load and improved pulmonary function, although to a lesser degree than the prophylactic regimen. When remdesivir was initiated 2 days post‐infection, however, it did not improve disease outcomes even though viral lung titers were reduced. The disease course in mice is accelerated compared to humans with viral titers peaking around day 2 concurrent with maximal damage to lungs.
The same group of investigators conducted a similar study evaluating remdesivir and lopinavir/ritonavir +/− interferon‐β against MERS‐CoV in an esterase‐deficient, humanized DDP4 mouse model. 19 Prophylactic remdesivir significantly improved MERS‐CoV–induced weight loss, decreased viral lung titers, and diminished features of acute lung injury compared to control mice. It also prevented mortality in mice administered a lethal dose of virus. In contrast, prophylactic lopinavir/ritonavir +/− interferon‐β slightly reduced viral loads but had no impact on other disease outcomes. Therapeutic remdesivir also led to improved disease outcomes and lower viral loads, but again, the effect size was diminished compared to prophylactic administration. Remdesivir administered after a lethal viral dose was given (as opposed to before with prophylaxis) did not prevent mortality. 19
The prophylactic and/or therapeutic efficacy of remdesivir has been evaluated in MERS‐CoV–infected and SARS‐CoV‐2–infected rhesus monkeys. These models more accurately recapitulate the lung disease observed in humans with mild‐moderate MERS‐CoV and SARS‐CoV‐2 infection compared to rodent models. 23 , 24 Dosing and pharmacokinetic analyses can also serve as a bridge to human dosing regimens. 7 However, due to the acute and accelerated infection course in monkeys, it is difficult to directly translate the timing of drug initiation to corresponding disease stages in humans. 23 , 24 Furthermore, neither model replicates many of the extrapulmonary disease manifestation seen in humans. 23 , 24
In the MERS‐CoV study, monkeys were administered remdesivir (5 mg/kg, which approximates drug exposures equivalent to 100 mg in humans) 24 hours before MERS‐CoV inoculation (prophylaxis) or 12 hours post‐inoculation (treatment). 8 Viral titers peak shortly after 12 hours in this model. For both groups, remdesivir was continued once daily until 6 days post‐inoculation. Prophylactic and therapeutic remdesivir treatment significantly reduced MERS‐CoV–induced clinical signs, viral titers in respiratory specimens, and the severity of lung lesions compared to control animals. These effects were most pronounced in the prophylactic group. 8 A similar prophylactic study was conducted using a higher remdesivir dose (10 mg/kg, which approximates drug exposures equivalent to 200 mg in humans). 14 Clinical signs and viral loads were significantly reduced vs. controls. Increases in serum creatinine and blood urea nitrogen suggestive of altered renal function were observed only in remdesivir‐treated animals. 14
In the SARS‐CoV‐2 study, remdesivir was again initiated shortly before viral titers are expected to peak at 12 hours post‐inoculation, and a dosing regimen equivalent to the regimen being tested in human COVID‐19 clinical trials was used (10 mg/kg load ~200 mg in humans, then 5 mg/kg daily ~100 mg daily in humans × 6 days). 7 Compared to vehicle‐treated monkeys, remdesivir‐treated monkeys had improved clinical and radiographic outcomes. Viral titers were reduced in lower respiratory tract specimens and undetectable at 3 days post inoculation. Viral titers were not reduced in upper respiratory tract specimens or in rectal swabs, however. 7 If translated in humans, the absence of a reduction in viral shedding in these sites may have implications for potential transmission risk following clinical improvement. 7
7. Clinical Data
Initial experience with remdesivir in patients with COVID‐19 emerged in the form of case reports and uncontrolled case series as detailed in Table 2. 3 , 25 , 26 , 27 , 28 , 29 All patients in these reports received remdesivir through compassionate use or expanded access programs. 14 , 30 The largest report included 61 patients from centers in North America, Europe, and Japan. 3 All patients were hospitalized with microbiologically confirmed COVID‐19 and had an oxygen saturation ≤ 94% on room air or required oxygen support. Those with severe renal impairment, elevated hepatic enzymes, or receiving another investigational agent at baseline were excluded. Of the 53 patients with short‐term follow‐up data available, 30 (56%) were receiving mechanical ventilation at baseline and a further 4 (8%) required extracorporeal membrane oxygenation (ECMO). Remdesivir was started at a median of 12 days following symptom onset. After a median follow‐up of 18 days, 7 (13%) patients died including 6 (18%) who were receiving invasive ventilation. Adverse events were reported in 32 (60%) patients, including 12 (23%) who experienced at least one serious adverse event (SAE). The most frequent adverse events were hepatic enzyme elevations, diarrhea, rash, renal impairment, and hypotension.
Table 2.
Case Reports and Case Series of Remdesivir Use in Patients with COVID‐19
| Reference | Study Design | Location | Patients Treated with Remdesivir | Time AFTER Symptom Onset of Remdesivir Initiation | Remdesivir Duration | Reported Adverse Effects After Starting Remdesivir | Outcomes at End of Follow‐up |
|---|---|---|---|---|---|---|---|
| Case series of 61 COVID‐19 patients | North America, Europe, and Japan |
N=61 34/53 receiving invasive ventilation at time of remdesivir initiation |
Median 12 days IQR 9–15 days |
40/53: 10 days 10/53: 5–9 days 3/53 < 5 days |
Any AE 32/53 Serious AE 12/53 12/53 hepatic enzyme increases 5/53 diarrhea 4/53 rash 4/53 renal impairment 4/53 hypotension |
7 no post day 1 data 1 erroneous remdesivir start date? 36/53 improvement in oxygen support category at median 18 days after remdesivir initiation 7/53 died in hospital (median 15 days after remdesivir initiation) |
|
| COVID‐19 Investigation Team 26 | Case series of 12 COVID‐19 patients | United States |
N=3 1/3 admitted to ICU at time of remdesivir initiation 0/3 required mechanical ventilation |
7–11 days | 4–10 days | GI AEs 3/3, Aminotransferase elevations in 3/3 patients | 3/3 symptoms resolved, 2/3 discharged home, 1/3 transferred to second healthcare facility |
| Lescure, et al. 28 | Case series of 5 COVID‐19 patients | France |
N=3 3/3 admitted to ICU at time of remdesivir initiation |
7–15 days | 5–10 days | 1/3 alanine aminotransferase elevation (3 × ULN), maculopapular rash leading to drug discontinuation | 2/3 symptoms resolved, 2/3 discharged home, 1/3 died in hospital 24 days after illness onset (10 days after re‐initiating remdesivir) |
| Hillaker et al. 25 | Case report | United States |
N=1 Admitted to ICU and invasive ventilation at time of remdesivir initiation |
13 days | 10 days | None | Extubated and stable in hospital |
| Sanville et al. 29 | Case report | United States |
N=1 Admitted to ICU and invasive ventilation at time of remdesivir initiation |
13 days | 10 days | None | Extubated and stable in hospital |
| Pereira et al. 27 | Case series of 90 solid organ transplant recipients with COVID‐19 | United States | N=2 | Not reported | Not reported | Not reported | Not reported |
AE = adverse event; N = number; ICU = intensive care unit; IQR = interquartile range; ULN = upper limit of normal.
The publication of this report generated a great deal of controversy. The authors rightly acknowledge many of its limitations including the small sample size, the short duration of follow‐up, missing data, and the absence of a control group. 3 The lack of post day 1 data on 7 patients is particularly concerning and not adequately addressed in the publication. In addition, a target sample size was not reported nor was a rationale given for why the data were analyzed and reported at the point they were. It is unclear how many physicians applied for compassionate use for their patients but were denied. It would be interesting to know how these patients differed from the small number whose request for remdesivir was approved. Finally, pre‐clinical studies suggest that remdesivir has little benefit when administered after the peak in viral replication has occurred. 7 , 8 , 9 , 14 , 19 Although the precise timing of peak viral loads was not accessed in these patients, it may be problematic to attribute favorable outcomes to the remdesivir when initiation was delayed beyond 12 days for many patients.
Against this backdrop, the first randomized, double‐blind, placebo‐controlled study evaluating remdesivir for COVID‐19 was published. 31 In this study, 237 hospitalized adults with severe COVID‐19 were enrolled at 10 centers in Wuhan, Hubei, China, and randomized (2:1) to remdesivir or matching placebo for a planned 10‐day treatment course (200 mg IV day 1, 100 mg IV daily on days 2 to 10). Severe COVID‐19 was defined as a SARS‐CoV‐2–positive respiratory specimen by RT‐PCR, and pneumonia was confirmed by chest imaging and an oxygen saturation ≤ 94% on room air or a ratio of arterial oxygen partial pressure to fractional inspired oxygen ≤ 300 mmHg. The primary outcome was time to clinical improvement defined as discharge alive from hospital or a 2‐point improvement on a 6‐point ordinal scale adapted from the World Health Organization’s 8‐point illness severity scale. 32 The study was stopped before reaching its target sample size of 453 due to slow enrollment.
Baseline characteristics were similar between groups: the median duration of illness before enrollment was 10 days and most patients (82%) required only low‐flow supplemental oxygen at baseline. 31 Use of antibiotics (91%) and corticosteroids (66%) were high in both groups. In addition, 28% and 32% of patients received lopinavir/ritonavir and/or interferon‐α2β, respectively during the course of their illness. With regards to the primary outcome, there was no significant difference in time to clinical improvement between the remdesivir and placebo groups [median 21 (interquartile range [IQR] 13–8) days vs. 23 (IQR 15–28), respectively; hazard ratio [HR] 1.23 (95% confidence interval [CI] 0.87–1.75)]. There were also no significant differences in key secondary endpoints including 28‐day mortality [remdesivir 14% vs. placebo 13%, difference 1.1% (95% CI −8.1‐10.3)]. The authors highlight signals that, among patients enrolled early (≤10 days after symptom onset), those in the remdesivir arm may have done marginally better than those in the placebo arm [time to clinical improvement 18 (IQR 12–28) days vs. 23 (IQR 15–28) days; 28‐day mortality 11% vs. 15%, difference – 3.6% (95% CI −16.2%–8.9%)], however this requires confirmation in future adequately powered clinical trials or meta‐analyses. Considering remdesivir’s potent in vitro activity against SARS‐CoV‐2 and impressive results in pre‐clinical models, one of the most unexpected findings in this study was that remdesivir had no impact on viral load. It is plausible that the delayed time to administration may have played a role. On the other hand, in vitro activity and animal data often do not translate into meaningful benefit for patients. 5 , 33 , 34
Strengths of this study include its randomized, double‐blind, placebo‐controlled design, high protocol adherence and low loss to follow‐up. 31 Premature termination however leaves an underpowered study with inconclusive results. Although cautious of over‐interpretation, the point estimate for the primary outcome (HR 1.23) suggests any benefit of remdesivir may be more modest than hoped for (HR 1.40) and the small difference in favor of remdesivir for the composite primary outcome appeared to be driven largely by change in oxygenation status rather than the more clinically meaningful hospital discharge. As discussed in the safety section, this study did give valuable data to help better characterize the adverse effect profile of remdesivir.
On the same day that the peer‐reviewed publication of the study by Wang and colleagues was released, preliminary results for the first stage of the highly anticipated National Institute of Allergy and Infectious Diseases (NIAID)‐sponsored Adaptive COVID‐19 trial (ACTT‐1) were announced in a press release 31 and the peer‐reviewed manuscript was published approximately 3 weeks laer. 35 ACTT‐1 was an adaptive, multicenter, randomized (1:1), double‐blind, placebo‐control trial evaluating remdesivir (200mg IV day 1, then 100mg IV days 2 to 10) in hospitalized adult patients with COVID‐19. Hospitalized adult patients with a SARS‐CoV‐2 RT‐PCR positive respiratory specimen and one of the following signs of a lower respiratory tract infection were eligible: radiographic infiltrates, a peripheral oxygen saturation ≤ 94% on room air or the need for respiratory support. The primary endpoint, was time to recovery within 28 days after randomization on an 8‐point ordinal scale and specifically recovery was defined as 1) not hospitalized and no limitations on activity; 2) not hospitalized with limitations on activity and/or need for supplemental oxygen; or 3) hospitalized but not requiring ongoing medical care for COVID‐19. The primary endpoint was initially defined as the difference in clinical status on the 8‐point ordinal scale between groups at day 15. However, as external data accumulated suggesting the clinical course of COVID‐19 was more protracted than originally anticipated, study statisticians (who were unaware of treatment assignments and outcomes) recommended the endpoint be changed approximately 3 weeks before the data cut‐off date for this preliminary analysis.
In total 1063 patients meet eligibility criteria and underwent randomization, of whom 731 (68.8%) had day 29 outcome data for the preliminary analysis. Baseline characteristics were well balanced between groups with the possible exception of need for mechanical ventilation or ECMO (remdesivir 23.1% versus placebo 28.2%). Most patients (79.8%) were enrolled from sites in North America and the majority (53.2%) were white. The median number of days from symptom onset to randomization was 9 and 88.7% had severe infection at baseline (category 5 on ordinal scale). 35
With regards to the primary outcome, time to recovery was shorter in the remdesivir group compared to the placebo group [11 days vs. 15 days; rate ratio (RR) 1.32 (95% CI 1.12 – 1.55)]. 35 The effect size was greatest in the group of patients requiring only supplemental oxygen at baseline [RR 1.47 (95% CI 1.17 – 1.84)], although the smaller numbers of patients in other groups precludes precise estimates of treatment effects. Furthermore, among patients requiring mechanical ventilation or ECMO at baseline, the median time to recovery was not reached at the data cut‐off date, suggesting longer follow‐up may be needed to evaluate the impact of remdesivir in the most critically ill patients. Interestingly, symptom duration prior to randomization did not appear to impact time to recovery [≤ 10 days RR 1.28 (95% CI 1.05 – 1.57) vs, > 10 days RR 1.38 (95% CI 1.05 – 1.81)]. Fourteen‐day mortality (n=1059) was numerically, but not significantly lower, in the remdesivir group [(7.1% vs. 11.6% (95% CI 0.47 – 1.04)]. Analysis of the data set with completed follow‐up to day 29 for all patients may yield additional insight although interpretation will be complicated by unblinding and crossover. Moving forward with ACTT‐2, remdesivir will be standard of care and subjects will be randomized to receive the Janus‐associated kinase inhibitor, baricitinib, or placebo. 36
In a press release, Gilead announced that similar outcomes were demonstrated in their open‐label, multicenter clinical trial of 5 vs. 10 days of remdesivir in patients with COVID‐19 not requiring invasive mechanical ventilation or ECMO. 37 More comprehensive data about this study is anticipated. If confirmed, this would allow the limited drug supply to treat many more patients. As of May 26, 2020, there are 10 clinical studies underway and registered on clinicaltrials.gov, including 6 randomized controlled trials, the full results of which are eagerly awaited. 38 (Table 3).
Table 3.
Ongoing Clinical Studies Registered on ClinicalTrials.gov of Remdesivir for COVID‐19 (adapted from 31 )
| ClinicalTrials.gov Identifier | Study Design | Intervention/Treatment of INTEREST | Location | Primary Outcome | Target Sample Size | Sponsor |
|---|---|---|---|---|---|---|
| NCT04365725 | Observational, retrospective, multicenter, cohort |
|
France | Clinical status on 7‐point ordinal scale at day 15 | 200 | Assistance Publique ‐ Hôpitaux de Paris |
| NCT04292899 | Randomized, multicenter, open‐label |
|
Multinational | Clinical status on 7‐point ordinal scale at day 14 | 6000 | Gilead |
| NCT0429730 | Randomized, multicenter, open‐label |
|
Multinational | Clinical status on 7‐point ordinal scale at day 11 | 1600 | Gilead |
| NCT04280705 | Adaptive, randomized, multicenter, double‐blind, placebo‐controlled |
|
Multinational | Time to recovery through day 29 according to 3‐point ordinal scale | 800 | National Institute of Allergy and Infectious Diseases (NIAID) |
| NCT04323761 | Observational, multicenter (expanded access program) |
|
Multinational | Not reported | Not reported | Gilead |
| NCT04330690 |
Adaptive, randomized, multicenter, open‐label Canadian arm of the WHO SOLIDARITY study |
|
Canada | All‐cause mortality at 29 days or at hospital discharge | 440 | Sunnybrook Health Sciences Centre |
| NCT04321616 |
Adaptive, randomized, multicenter, open‐label Norwegian arm of the WHO SOLIDARITY study |
|
Norway | In hospital mortality at 21 days | 700 | Oslo University Hospital |
| NCT04302766 | Observational, multicenter (expanded access program) |
|
Not reported | Not reported | Not reported | U.S. Army Medical Research and Development Command |
| NCT04315948 | Adaptive, randomized, multicenter, open‐label |
|
France | Clinical status on 7‐point ordinal scale at day 15 | 3100 | Institut National de la Santé Et de la Recherche Médicale |
| NCT04373044 | Prospective, 2‐center, single‐arm |
|
USA | All‐cause mortality or need for mechanical ventilation up to day 14 | 59 | University of Southern California |
8. Adverse Effects
Information on the safety profile of remdesivir is rapidly evolving. Until recently, most clinical experience has been in patients with Ebola virus infection, which has very different clinical manifestations compared to COVID‐19, making extrapolation of drug safety across populations problematic. 5 , 14 In the PALM study, 9 of 175 patients who received remdesivir for Ebola virus infection experienced SAEs judged by trial investigators to be potentially related to remdesivir. 5 The most serious of these was hypotension during the loading dose rapidly followed by cardiac arrest and death. 5 Among 38 Ebola virus infection survivors who were enrolled in the single‐arm phase II PREVAIL IV study, 1 patient required a remdesivir dose reduction due to transaminase elevations. 14
Safety data from four phase 1 PK studies in healthy volunteers have also been partly reported. 3 , 23 In these studies, subjects received single remdesivir doses of up to 225 mg or multiple doses of 150 mg once daily for 7 or 14 days or 200 mg once followed by 100 mg daily for a total of 5 or 10 days. The most common adverse events, recorded in at least five subjects, were phlebitis, constipation, headache, ecchymosis, nausea and extremity pain. 3 Transient asymptomatic grade 1 or 2 alanine aminotransferase (ALT) elevations were observed in most subjects (exact frequency not reported) in the multi‐dose PK studies including one individual with ALT values> 10 × baseline. 3 , 23
Transaminase increases have also been reported in COVID‐19 patients treated with compassionate use remdesivir (Table 2) 3 , 23 , 26 , 28 In the report detailing the first 12 COVID‐19 cases in the US, all three patients who received remdesivir experienced transient transaminase elevations and gastrointestinal symptoms. 26 In addition one patient in the case series from France experienced ALT elevation to 3 times the upper limit of normal and a maculopapular rash leading to remdesivir discontinuation 4 days following the first dose. 28 The FDA reports that among 163 patients enrolled in the compassionate use program, the overall incidence of liver function test abnormalities was 11.7%. 23 Seven cases were considered SAEs. The time to onset from the first dose ranged from 1 to 16 days. With the exception of 1 case of elevated bilirubin, none of the other cases had associated hyperbilirubinemia or symptoms of hepatitis. 23
In an RCT, adverse effects were recorded in 66% of patients randomized to remdesivir of which 8% were grade 3 or 4 (thrombocytopenia n=4, hypokalemia n=2, hyperkalemia n=2, anemia n=1, increased total bilirubin n=1). 31 The incidence and distribution of adverse effects were similar in the placebo group. Aspartate aminotransferase (AST) elevations were observed in 5% of patients in the remdesivir arm compared to 12% in the placebo arm. No patients in either arm experienced grade 3 or 4 transaminase elevations. More patients in the remdesivir discontinued treatment prematurely (12% vs. 5%) with respiratory failure or acute respiratory distress syndrome being the most common event leading to drug discontinuation in the remdesivir group. In a summary of safety data reported by the FDA from the a remdesivir clinical trial comparing 5‐ and 10‐day treatment courses in patients with COVID‐19, grade 3 and 4 ALT and/or AST elevations occurred in 7% patients. Elevations in bilirubin were uncommon (1.3%). 23
No new safety signals were detected in the ACTT‐1 study. 35 SAEs were reported in 21.1% patients in the remdesivir group compared to 27.0% in the placebo group with the most commonly reported in both groups being respiratory failure (remdesivir 5.2% versus placebo 8.0%) and hypoxia / respiratory distress (remdesivir 2.4% versus placebo 2.8%). The incidence and distribution of grade 3 or 4 adverse events were also similar in the 2 groups (overall remdesivir 28.5% versus placebo 33.0%, respectively). Increased aminotransferase levels occurred in 4.1% of the remdesivir group compared to 5.9% of the placebo group.
At this time, it is unclear if the liver enzyme abnormalities seen in some patients receiving remdesivir for COVID‐19 are a component of the infectious process or due to the drug. Increased aminotransferases were infrequent and occurred in similar proportions of remdesivir and placebo treated patients in the RCT by Wang et al, and the ACTT‐1 study. 31 , 35 Although this is reassuring, the fact that abnormalities, albeit non‐severe, were seen in healthy volunteers suggests remdesivir was at least partly culpable. Whether asymptomatic abnormalities are harbingers of more serious liver toxicity is also unknown. Other approved nucleoside analogues, including those used to treat HIV, hepatitis B, and cytomegalovirus, are known to cause liver injury by a variety of mechanisms. 24 The most common involves inhibition of mitochondrial DNA synthesis leading to mitochondria depletion or dysfunction. First‐generation HIV reverse transcriptase inhibitors are thought to cause hepatic injury by this mechanism. Mitochondrial dysfunction can affect multiple tissues manifesting as myopathy, neuropathy, pancreatitis, bone marrow suppression, and/or hepatic injury. 24 Extra‐hepatic manifestations of mitochondrial dysfunction have not been reported in patients exposed to remdesivir to date. Nucleoside analogues may also cause liver injury through acute hypersensitivity reactions or the production of toxic intermediates. 24 These reactions tend to be idiosyncratic and uncommon, whereas transaminase elevations are consistently observed in a minority of remdesivir‐treated patients. A fulsome safety assessment of remdesivir will require thorough review of data from recently completed and ongoing studies in addition to post‐marketing surveillance and real‐world experience.
9. Drug Interactions
At the time of writing, no in vivo drug interaction studies of remdesivir have been published but the ability of remdesivir to inhibit or induce cytochrome P450 (CYP450) enzymes and transporters has been tested in vitro. 14 Importantly however, as a pro‐drug, remdesivir is rapidly degraded in vivo so the potential for clinically significant drug interactions is likely limited. 14 Data on the potential for remdesivir metabolites to perpetrate drug interactions are even scarcer. In in vitro studies, remdesivir was a weak inhibitor of CYP1A2, CYP2C9, CYP2C19, and CYP2D6. 14 Remdesivir’s IC50 for CYP3A was 1.6 μmol/L, suggesting inhibition may occur briefly with standard human exposures. Inhibition of CYP450 enzymes by metabolites was not investigated. 14 Tests of remdesivir CYP450 induction have been inconsistent; it may induce CYP1A2 and CYP2B6. 14 Again the clinical importance of this is questionable. GS‐441524 and GS‐704277 demonstrated no CYP450 induction in these studies. Remdesivir was found to be a substrate (OATP1B, P‐glycoprotein) or inhibitor (OAT1B1, OAT1B3) of several drug transporters. 14 There are no exclusion criteria related to drug‐drug interactions in current remdesivir clinical studies (Table 3).
10. Dosage and Administration
Remdesivir is available in two bioequivalent formulations: a concentrated solution (5 mg/mL) and a lyophilized powder formulation. 14 , 18 Vials contain 100 mg of remdesivir and are preservative free. 14 , 18 Readers are referred to the FDA Fact Sheet for full storage, preparation, and administration instructions. 18
For adults and children weighing ≥ 40 kg requiring invasive mechanical ventilation or ECMO, the recommended dose is 200 mg IV on day 1 followed by 100 mg IV once daily on days 2 to 10. For those not requiring invasive mechanical ventilation or ECMO, a 5‐day regimen is recommended. Doses should be administered over 30 minutes to 2 hours. Readers are referred to the FDA Fact Sheet for full pediatric dosing recommendations. 18 There is no information on direct IV push, intramuscular, or subcutaneous administration at this time.
11. Conclusions
At this time there are no therapies that have been scientifically proven to improve mortality in COVID‐19. Current management is largely focused on supportive care and prevention of complications. 39 , 40 Efficacious and safe antiviral agents are therefore urgently needed to relieve the burden on health‐care systems. As detailed in this review, remdesivir is a nucleoside analogue pro‐drug with unique structural features that allow high concentrations of the active triphosphate metabolite to be delivered intracellularly. 6 It evades proofreading to successfully inhibit viral RNA synthesis and has demonstrated potent antiviral activity against β‐coronaviruses, including SARS‐CoV‐2 both in vitro and in animal models. 7 , 8 , 9 , 10 , 11 , 12 , 15 , 19 , 20 These data, coupled with early safety data from clinical experience in Ebola virus infection, 5 provide strong rationale for prioritizing testing of remdesivir in COVID‐19 clinical trials. The unpredictable nature of a pandemic however poses many challenges to researchers attempting to conduct clinical trials. 41 As of April 30, 2020, more than 2000 patients with COVID‐19 have received remdesivir through compassionate use or expanded access programs. 37 It is impossible to know if these patients benefited or were harmed but we do know these programs do little to advance science. When patient enrollment in clinical trials is not available, many clinicians face intense pressure to offer unproven therapies based on compelling pre‐clinical data. Efforts must focus on ensuring the necessary infrastructure is in place to expand patient access to pragmatic clinical trials and make it simple for clinicians to enroll them. This could obviate the need for compassionate use programs.
Although the first randomized controlled trial evaluating remdesivir for COVID‐19 was conducted a multiple sites in the initial outbreak epicenter, it failed to meet its target sample size due to slow enrollment after the surge in cases diminished and produced inconclusive results. It did however provide data on the use of remdesivir gathered in a rigorous manner. ACTT‐1 represents a remarkable global effort with a total of 60 study sites in 10 countries enrolling more than 1000 patients over approximately 2 months. 35 Remdesivir treatment resulted in an accelerated time to recovery by 4 days which represents meaningful progress for patients and healthcare systems. The stubbornly high mortality rates and apparent absence of benefit among the most critically ill patients however suggests the need for more effective and / or adjunctive therapies. With at least 6 remdesivir randomized‐controlled trials currently underway worldwide with and without adjunctive immunomodulatory agents, there is reason to be optimistic that we will accumulate good data to more precisely define remdesivir’s therapeutic niche in COVID‐19.
Conflict of interest: SCJJ and RK have nothing to disclose. LDD has received conference development and administration support from Avir Pharma, Sunovion and Merck unrelated to this work. No funding was received for this work.
References
- 1. Sanders JM, Monogue ML, Jodlowski TZ, Cutrell JB. Pharmacologic treatments for coronavirus disease 2019 (COVID‐19): a review. JAMA 2020;323:1824–36 [DOI] [PubMed] [Google Scholar]
- 2. Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID‐19) outbreak in China: summary of a report of 72314 cases from the Chinese Center for Disease Control and Prevention. JAMA 2020;323:1239. [DOI] [PubMed] [Google Scholar]
- 3. Grein J, Ohmagari N, Shin D, et al. Compassionate use of remdesivir for patients with severe Covid‐19. N Engl J Med 2020;382:2327–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McCreary EK, Pogue JM. Coronavirus disease 2019 treatment: a review of early and emerging options. Open Forum Infect Dis 2020;7:ofaa105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mulangu S, Dodd LE, Davey RT Jr, et al. A randomized, controlled trial of Ebola virus disease therapeutics. N Engl J Med 2019;24:2293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Siegel D, Hui HC, Doerffler E, et al. Discovery and synthesis of a phosphoramidate prodrug of a Pyrrolo[2,1‐f][triazin‐4‐amino] adenine C‐nucleoside (GS‐5734) for the treatment of Ebola and emerging viruses. J Med Chem 2017;5:1648–61. [DOI] [PubMed] [Google Scholar]
- 7. Williamson BN, Feldmann F, Schwarz B, et al. Clinical benefit of remdesivir in rhesus macaques infected with SARS‐CoV‐2. bioRxiv preprint 10.1101/2020.04.15.043166 Posted 15 April 2020. [DOI] [PMC free article] [PubMed]
- 8. de Wit E, Feldmann F, Cronin J, et al. Prophylactic and therapeutic remdesivir (GS‐5734) treatment in the rhesus macaque model of MERS‐CoV infection. Proc Natl Acad Sci USA 2020;12:6771–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sheahan TP, Sims AC, Graham RL, et al. Broad‐spectrum antiviral GS‐5734 inhibits both epidemic and zoonotic coronaviruses. Sci Transl Med 2017;9:eaal3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gordon CJ, Tchesnokov EP, Woolner E, et al. Remdesivir is a direct‐acting antiviral that inhibits RNA‐dependent RNA polymerase from severe acute respiratory syndrome coronavirus 2 with high potency. J Biol Chem 2020;295:6785–6797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Agostini ML, Andres EL, Sims AC, et al. Coronavirus susceptibility to the antiviral remdesivir (GS‐5734) is mediated by the viral polymerase and the proofreading exoribonuclease. MBio 2018;9:e00221–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gordon CJ, Tchesnokov EP, Feng JY, Porter DP, Gotte M. The antiviral compound remdesivir potently inhibits RNA‐dependent RNA polymerase from Middle East respiratory syndrome coronavirus. J Biol Chem 2020;15:4773–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shannon A, Le NT, Selisko B, et al. Remdesivir and SARS‐CoV‐2: Structural requirements at both nsp12 RdRp and nsp14 Exonuclease active‐sites. Antiviral Res 2020;178:104793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. European Medicines Agency . Human Medicines Division. Summary on compassionate use. Remdesivir Gilead. Product No. EMEA/H/K/5622/CU. 03 April 2020. https://www.ema.europa.eu/en/documents/other/summary‐compassionate‐use‐remdesivir‐gilead_en.pdf Accessed 04/24/2020
- 15. Wang M, Cao R, Zhang L, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019‐nCoV) in vitro. Cell Res 2020;3:269–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Warren TK, Jordan R, Lo MK, et al. Therapeutic efficacy of the small molecule GS‐5734 against Ebola virus in rhesus monkeys. Nature 2016;7594:381–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kiser TH, Fish DN, Aquilante CL, et al. Evaluation of sulfobutylether‐beta‐cyclodextrin (SBECD) accumulation and voriconazole pharmacokinetics in critically ill patients undergoing continuous renal replacement therapy. Crit Care 2015;19:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fact sheet for health care providers emergency use authorization (EUA) of remdesivir (GS‐5734™). May 1, 2020 https://www.fda.gov/media/137566/download
- 19. Sheahan TP, Sims AC, Leist SR, et al. Comparative therapeutic efficacy of remdesivir and combination lopinavir, ritonavir, and interferon beta against MERS‐CoV. Nat Commun 2020;1:222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brown AJ, Won JJ, Graham RL, et al. Broad spectrum antiviral remdesivir inhibits human endemic and zoonotic deltacoronaviruses with a highly divergent RNA dependent RNA polymerase. Antiviral Res 2019;169:104541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Choy KT, Wong AY, Kaewpreedee P, et al. Remdesivir, lopinavir, emetine, and homoharringtonine inhibit SARS‐CoV‐2 replication in vitro. Antiviral Res 2020;178:104786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chang S, Zhuang D, Guo W, et al. The antiviral activity of approved and novel drugs against HIV‐1 mutations evaluated under the consideration of dose‐response curve slope. PLoS One 2016;3:e0149467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Munster VJ, de Wit E, Feldmann H. Pneumonia from human coronavirus in a macaque model. N Engl J Med 2013;16:1560–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Munster VJ, Feldmann F, Williamson BN, et al. Respiratory disease and virus shedding in rhesus macaques inoculated with SARS‐CoV‐2. bioRxiv preprint 10.1101/2020.03.21.001628bioRxiv. Posted 21 March 2020. [DOI] [PMC free article] [PubMed]
- 25. Hillaker E, Belfer JJ, Bondici A, Murad H, Dumkow LE. Delayed initiation of remdesivir in a COVID‐19 positive patient. Pharmacotherapy 2020;40:592–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Team C‐I . Clinical and virologic characteristics of the first 12 patients with coronavirus disease 2019 (COVID‐19) in the United States. Nat Med 2020; 10.1038/s41591-020-0877-5 [DOI] [PubMed] [Google Scholar]
- 27. Pereira MR, Mohan S, Cohen DJ, et al. COVID‐19 in Solid Organ Transplant Recipients: Initial Report from the US Epicenter. Am J Transplant 2020. 10.1111/ajt.15941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lescure FX, Bouadma L, Nguyen D, et al. Clinical and virological data of the first cases of COVID‐19 in Europe: a case series. Lancet Infect Dis 2020;20:697–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sanville B, Corbett R, Pidcock W, et al. A community transmitted case of severe acute respiratory distress syndrome due to SARS CoV2 in the United States. Clin Infect Dis 2020. 10.1093/cid/ciaa347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Emergency access to remdesivir outside of clinical trials. Gilead global protal. https://www.gilead.com/purpose/advancing‐global‐health/covid‐19/emergency‐access‐to‐remdesivir‐outside‐of‐clinical‐trials
- 31. Wang Y, Zhang D, Du G, et al. Remdesivir in adults with severe COVID‐19: a randomised, double‐blind, placebo‐controlled, multicentre trial. Lancet 2020;395:1569–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. World Health Organization . Covid‐19 therapeutic trial synopsis. 18 Feb. 2020 https://www.who.int/publications‐detail/covid‐19‐therapeutic‐trial‐synopsis
- 33. Harding JD. Nonhuman primates and translational research: progress, opportunities, and challenges. ILAR J 2017;2:141–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sperber K, Louie M, Kraus T, et al. Hydroxychloroquine treatment of patients with human immunodeficiency virus type 1. Clin Ther 1995;4:622–36. [DOI] [PubMed] [Google Scholar]
- 35. Beigel JH, Tomashek KM, Dodd LE, et al. Remdesivir for the Treatment of Covid‐19 ‐ Preliminary Report. N Engl J Med 2020. https://doi,org/10.1056/NEJMoa2007764 [DOI] [PubMed] [Google Scholar]
- 36. National Institutes of Health. US Department of Health and Human Services. News release. NIH clinical trial testing antiviral remdesivir plus anti‐inflammatory drug baricitinib for COVID‐19 begins.. 2020. https://www.nih.gov/news‐events/news‐releases/nih‐clinical‐trial‐testing‐antiviral‐remdesivir‐plus‐anti‐inflammatory‐drug‐baricitinib‐covid‐19‐begins. Accessed 26 May 2020.
- 37. Gilead Q1 2020 Earnings Results. April 30, 2020. http://investors.gilead.com/static‐files/af4599eb‐4fb8‐4cf7‐96a1‐38caf477e9b4
- 38. https://clinicaltrials.gov/ct2/results?cond=COVID‐19+or+SARS‐CoV‐2&term=remdesivir+&cntry=&state=&city=&dist Accessed 5 May 2020.
- 39. Fowler R, Hatchette T, Salvadori M, et al. Clinical Management of Patients with Moderate to Severe COVID‐19 ‐ Interim Guidance. Public Health Agency of Canada/Canadian Critical Care Society/Association of Medical Microbiology and Infectious Disease Canada. April 2020. https://www.ammi.ca/Content/Clinical%20Care%20COVID‐19%20Guidance%20FINAL%20April2%20ENGLISH%281%29.pdf. Accessed 04/16/2020
- 40. Bhimraj A, Morgan RL, Shumaker AH, et al. Infectious diseases society of America guidelines on the treatment and management of patients with COVID‐19. Clin Infect Dis 2020; 10.1093/cid/ciaa478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Muller MP, McGeer A, Straus SE, Hawryluck L, Gold WL. Clinical trials and novel pathogens: lessons learned from SARS. Emerg Infect Dis 2004;3:389–94. [DOI] [PMC free article] [PubMed] [Google Scholar]