

Abstract
Virus invasion triggers host immune responses, in particular, innate immune responses. Pathogen‐associated molecular patterns of viruses (such as dsRNA, ssRNA, or viral proteins) released during virus replication are detected by the corresponding pattern‐recognition receptors of the host, and innate immune responses are induced. Through production of type‐I and type‐III interferons as well as various other cytokines, the host innate immune system forms the frontline to protect host cells and inhibit virus infection. Not surprisingly, viruses have evolved diverse strategies to counter this antiviral system. In this review, we discuss the multiple strategies used by proteases of positive‐sense single‐stranded RNA viruses of the families Picornaviridae, Coronaviridae, and Flaviviridae, when counteracting host innate immune responses.
Keywords: cleavage of host proteins, innate immunity, viral protease
Abbreviations
ADNP, activity‐dependent neuroprotective protein
BVDV, bovine viral diarrhea virus
CSFV, classical swine fever virus
DENV, dengue virus
DUB, deubiquitination
HCV, hepatitis C virus
IFN, interferon
ISG, IFN‐stimulated gene
MERS‐CoV, Middle‐East respiratory syndrome coronavirus
Mpro, main protease
pDCs, plasmacytoid dendritic cells
PLpro, papain‐like protease
SARS‐CoV, severe acute respiratory syndrome coronavirus
TBEV, tick‐borne encephalitis virus
WNV, West Nile virus
YFV, yellow fever virus
Proteases of positive‐sense single‐stranded RNA ((+)ssRNA) viruses
Since about two decades, emerging and re‐emerging viruses have caused an increasing number of disease outbreaks in humans. In particular, emerging RNA viruses pose great threats to public health, for example, Ebola virus, Zika virus (ZIKV), and Middle‐East respiratory syndrome coronavirus (MERS‐CoV). The latter two are positive‐sense single‐stranded RNA ((+)ssRNA) viruses. We will briefly introduce the proteases of three (+)ssRNA virus families here, namely Picornaviridae, Coronaviridae, and Flaviviridae. This will be followed by a description of the complex signaling pathways that lead from recognition of the viral intruders to the production of antiviral cytokines, such as type‐I and type‐III interferons (IFNs). Finally, we will discuss the mechanisms by which the viral proteases interfere with these signaling pathways.
The Picornaviridae family includes a large number of small nonenveloped (+)ssRNA viruses with a genome size between 7.5 and 10 kb. As of March 2017, this family contains 35 genera with 80 species 1. The genera Enterovirus, Hepatovirus, Aphthovirus, and Cardiovirus have been well investigated. Picornaviruses can cause several severe diseases in man and animals, such as poliomyelitis, hepatitis, and encephalitis. The viral genome usually encodes a polyprotein comprising regions P1, P2, and P3 (Fig. 1A) 2, 3. The P1 region includes the structural proteins, while the latter two comprise nonstructural proteins, including the enzymes required for polyprotein processing and RNA replication. P1, P2, and P3 are further cleaved by viral proteases into mature proteins. P1 is digested to 1A (also known as VP4), 1B (VP2), 1C (VP3), and 1D (VP1); P2 is processed to 2A, 2B, and 2C; whereas P3 becomes 3A, 3B, 3C, and 3D 2, 3. Picornaviruses encode up to three proteases, the 2A protease (2Apro), the 3C protease (3Cpro), and – in case of some family members (e.g. the genera Aphthovirus and Cardiovirus) – the leader protease (Lpro) (Fig. 1A) 4.
Figure 1.
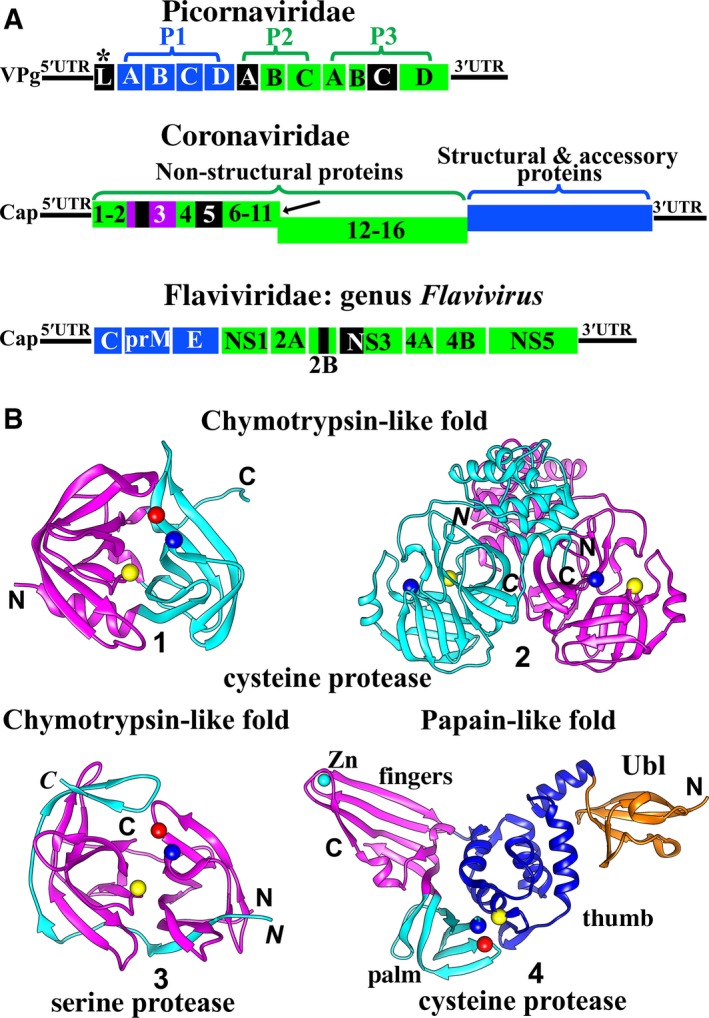
(A) Genome organization of picornaviruses, coronaviruses, and flaviviruses. All structural and accessory proteins are shown in blue. Nonstructural proteins are shown in green, with the exception of proteases, which are shown in black. Picornaviridae : The 5′ end of the picornavirus genomic RNA is covalently bound to VPg (viral protein genome‐linked). The genome encodes a polyprotein comprising the three regions P1 (structural proteins), P2, and P3 (nonstructural proteins). Generally, two viral proteases, 2Apro and 3Cpro, cleave the polyprotein into mature proteins. P1 is processed to yield 1A (VP4), 1B (VP2), 1C (VP3), and 1D (VP1); P2 is processed to 2A, 2B, and 2C; while P3 cleavage products are 3A, 3B, 3C, and 3D. *In some picornavirus genera (e. g. Aphthovirus, Cardiovirus), a third viral protease exists, the leader protease (Lpro). It auto‐cleaves itself from the polyprotein. Coronaviridae:CoVs possess the largest genome of all known RNA viruses. The 5′ genomic RNA carries a methylated cap. Two open‐reading frames (ORFs), 1a and 1b, occupy the 5′‐terminal two thirds of the CoV genome. ORF1a encodes the polyprotein 1a (Nsp1‐11), while ORF1a plus ORF1b are translated into the polyprotein 1ab (Nsp1‐16); this involves a (‐1) ribosomal frameshift overreading the stop codon of ORF1a (indicated by the black arrow). The 3′‐proximal third encodes the structural and accessory proteins. The polyproteins pp1a and pp1ab are processed by the viral proteases PL pro (within Nsp3; Nsp3 is purple) and Mpro (3CL pro, Nsp5). The genomes of members of the Flaviviridae differ between genera. Here, a genome of a member of the genus Flavivirus is shown as an example. The 5′‐capped genome encodes a polyprotein, which is cleaved into three structural proteins as well as seven nonstructural proteins by host and viral proteases. Flaviviruses have only one protease, the NS2B/NS3pro. NS2B is a cofactor for the NS3 serine protease. (B) Structures of proteases of +ssRNA viruses. The fold of most RNA‐virus proteases belongs to either the chymotrypsin‐like class or the papain‐like class. The chymotrypsin fold consists of two β‐barrel domains, while the typical papain‐like fold contains an α‐helical domain and a predominantly β‐sheet domain. The catalytic residues are located in the cleft between the two domains in both chymotrypsin‐like and papain‐like proteases. Picornavirus 2Apro, 3Cpro, coronavirus 3CL pro (Mpro), HCV and pestivirus NS3/NS4Apros, as well as flavivirus NS2B/NS3pro, adopt the chymotrypsin‐like fold, whereas picornavirus Lpro and coronavirus PL pro feature the papain‐like fold. 1) The structure of enterovirus D68 3Cpro 19 (PDB entry: 3ZV8). The Cα atoms of the catalytic triad Cys–His–Glu are shown as yellow, blue, and red spheres, respectively. 2) The structure of transmissible gastroenteritis virus (TGEV, a CoV) 3CL pro (Mpro) 24 (PDB entry: 1LVO). Dimerization of the 3CL pro (Mpro) is a prerequisite for its activity. The two protomers are displayed in cyan and purple. The catalytic dyad Cys–His (Cα atoms shown as yellow and blue spheres) is located within the chymotrypsin‐like subdomain of each monomer. An additional α‐helical domain also exists in each protomer. 3) The structure of Zika virus NS2B/NS3pro 22 (PDB entry 5LC0). The NS3 protease is shown in purple and the NS2B cofactor is in cyan. The Cα atoms of the catalytic triad Ser–His–Asp are shown as yellow, blue, and red spheres, resp. 4, The structure of MERS‐CoV PL pro 26 (PDB entry 4P16). In the coronavirus PL pro, the β‐sheet domain is larger than in the canonical papain‐like fold and divided into two subdomains, fingers (purple) and palm (cyan); together with the thumb subdomain (α‐helical domain; blue), an extended right‐hand fold is the result. A ubiquitin‐like (Ubl) domain (orange) is located in the N‐terminal region of the PL pro. The Cα atoms of the catalytic triad residues Cys–His–Asp are shown as yellow, blue, and red spheres, resp. All figures in (B) have been prepared by using UCSF Chimera 183.
The Coronaviridae family is divided into two subfamilies, Coronavirinae and Torovirinae 1. Two recently emerged human coronaviruses from the subfamily Coronavirinae, severe acute respiratory syndrome coronavirus (SARS‐CoV) and MERS‐CoV, can cause severe pneumonia. In particular, the latter virus frequently also leads to renal failure 5. Coronaviruses are enveloped +ssRNA viruses and have the largest genome (26–32 kb) of all known RNA viruses. The 5′‐terminal two thirds of the genome contain the two open‐reading frames (ORFs) 1a and 1b. ORF1a codes for polyprotein 1a containing nonstructural protein 1–11 (Nsp1–11), while ORF1a and ORF1b together encode polyprotein 1ab comprising Nsp1–16. This latter mechanism features a (‐1) ribosomal frameshift overreading the stop codon of ORF1a (Fig. 1A) 6. The 3′‐proximal third encodes the structural and accessory proteins 7, 8. These two polyproteins are processed into 15 or 16 mature Nsps to form the replication/transcription complex. This step is performed by two types of viral proteases, namely, one or two papain‐like proteases (PLpro(s)) located within Nsp3, and a main protease (Mpro) (Nsp5), which is frequently also called ‘3C‐like protease’ (3CLpro) (Fig. 1A; see 9 for a review).
The family Flaviviridae includes four genera: Hepacivirus, Flavivirus, Pestivirus, and Pegivirus. Here, we discuss viral proteases from the former three genera. Since the genome organization and the proteases are different in these three genera of the Flaviviridae family, we will introduce them separately.
Viruses from the genus Hepacivirus are enveloped (+)ssRNA viruses. The best characterized member of this genus is hepatitis C virus (HCV). This virus can lead to acute and chronic hepatitis. About 71 million people have chronic hepatitis C infection worldwide (http://www.who.int, last accessed on August 16, 2017). The genome of HCV is about 9.6 kb in size and encodes a polyprotein precursor that is processed by host and two viral proteases to yield four structural (C, E1, E2, and p7) and six nonstructural proteins (NS2, NS3, NS4A, NS4B, NS5A, and NS5B). These two viral proteases are the NS2 autoprotease (NS2pro) and the NS3/NS4A protease (NS3/NS4Apro) 10, 11. NS4A is a cofactor for the NS3 protease. In HCV, the latter enzyme is also called NS3/4Apro in many publications; however, we will use ‘NS3/NS4Apro’ in what follows, in order to be consistent with the NS2B/NS3pro notation in the flaviviruses.
Viruses of the genus Flavivirus are also enveloped (+)ssRNA viruses. Members of the genus include dengue virus (DENV), West Nile virus (WNV), yellow fever virus (YFV), tick‐borne encephalitis virus (TBEV), Zika virus (ZIKV), etc. Flaviviruses are mainly transmitted by arthropods, such as mosquitoes or ticks. Many of the mosquito‐borne family members are highly endemic in the tropics and subtropics, whereas TBEV is prevalent in Central and Eastern Europe. The ~11‐kb genome of flaviviruses encodes a polyprotein that is cleaved into three structural (C, prM, and E) and seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) by host and viral proteases (Fig. 1A). The genus Flavivirus only has one protease, NS2B/NS3pro 12.
Viruses of the genus Pestivirus mainly infect mammals, such as cattle and swine. Bovine viral diarrhea virus (BVDV) and classical swine fever virus (CSFV) belong to this genus. The genome of pestiviruses encodes a polyprotein that is processed by viral and host proteases into 12 mature proteins (Npro, C, Erns, E1, E2, p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B). In this genus, three proteolytic enzymes, the N‐terminal protease (Npro), the NS2pro, and the NS3/NS4Apro, have been identified 13, 14, 15.
Although the primary structures of proteases in these three (+)ssRNA virus families are very different from one another, the three‐dimensional structures of most proteases belong to either the chymotrypsin‐like fold or the papain‐like fold (Fig. 1B). The typical chymotrypsin fold consists of two β‐barrel domains, each containing six β‐strands. The catalytic residues are located in the cleft between these two domains. Picornavirus 2Apro 16, 3Cpro 17, 18, 19, HCV NS3/NS4Apro 20, flavivirus NS2B/NS3pro 21, 22, and pestivirus NS3/NS4Apro 23 adopt this fold; however, the former two enzymes are cysteine proteases, while the latter three are serine proteases. The coronavirus Mpro (3CLpro) also possesses a chymotrypsin‐like fold, but with an additional α‐helical domain; furthermore, dimerization of this cysteine protease is a prerequisite for its activity (Fig. 2B) 9, 24. However, the picornavirus Lpro 4, 25 and the coronavirus PLpro 9, 26, 27 feature a papain‐like fold. The canonical papain‐like fold contains an α‐helical and a predominantly β‐sheet domain, with the active site located in the cleft between them. This is exactly what is found in the picornavirus Lpro 4, 25, whereas in the coronavirus PLpro, the β‐sheet domain is larger and further divided into two subdomains: fingers and palm. Together with the thumb subdomain (α‐helical domain), they form an extended right‐hand fold. In addition, a ubiquitin‐like (Ubl) domain is located at the N terminus of the PLpro (Fig. 1B) 9, 26, 27.
Figure 2.

Schematic overview of host innate immune pathways and their disruption by proteases of RNA viruses. The actions of viral proteases of three (+)ssRNA virus families, namely Picornaviridae, Coronaviridae, and Flaviviridae, are illustrated by triangle and square symbols. Triangles indicate cleavage of the target protein, while squares symbolize interaction with this protein in the absence of cleavage. Blue spheres indicate phosphorylation, and (Ub)x means polyubiquitination. (A) The TLR (Toll‐like receptor), RLR (retinoic acid‐inducible gene‐I (RIG‐I)‐like receptor), and NLR (nucleotide‐binding oligomerization domain (NOD)‐like receptor) signaling pathways. TLR7 detects ssRNA and triggers downstream signaling via the adaptor, MyD88 (myeloid differentiation primary response gene 88). Subsequently, MyD88 recruits IRAK4 (interleukin‐1 receptor‐associated kinase 4) to activate IRAK1/2, then IRAKs dissociate from MyD88 and bind to TNF receptor‐associated factor 6 (TRAF6). TRAF6 activates TAK1 (TGF‐β‐activated kinase 1). TAK1 further recruits TAB1/2/3 (TAK1‐binding protein 1/2/3), to activate IKKα/β/γ (IκB kinase alpha, beta, and gamma; IKKγ is also named NEMO). Then, the IKKs mediate the phosphorylation of IκB, the NF‐κB inhibitor. Phosphorylated IκB is degraded and releases NF‐κB to induce production of TNFs (tumor necrosis factors) and other cytokines. This pathway thus comprises TLR7→MyD88→IRAK4/1/2→TRAF 6→TAK1/TAB1–3→IKKα/β/γ→IκB→NF‐κB. Also, TRAF6, IRAK4, TRAF3, IKKα, and IRAK1 form a complex. In this complex, both IKKα and IRAK1 activate the IRF7 (interferon regulatory factor 7) but not the IRF3 pathway (see red arrows). TLR3 detects dsRNA and triggers TRAF3 and TRAF6 by the mediator, TRIF (TIR domain‐containing adaptor protein‐inducing IFNβ). TRAF3 activates the TBK1/IKKε (TANK‐binding kinase 1/IκB kinase epsilon)‐mediated IRF3/7 pathway. TANK (TRAF family member‐associated NF‐κB activator) and IKKγ can activate TBK1/IKKε. TBK1/IKKε further stimulate the IRF3/7 pathway. In addition, STING (stimulator of interferon genes) can upregulate IRF3 signaling. The main cascade of this pathway thus comprises TLR3→TRIF→TRAF 3→TBK1/IKKε→IRF3/7. TRAF6 and TRAF3 are typed in bold to indicate that they are located at central positions of pathways. The downstream cascade of TRAF6 activating the NF‐κB pathway is the same as for the TLR7 pathway. RLRs detect ssRNA/dsRNA and trigger the activation of TRAF3 and TRAF6 by the mediator, MAVS (mitochondrial antiviral‐signaling protein; also known as IPS‐1, Cardif, VISA). The downstream signaling pathway is the same as for the TLR3 pathway. NLRs include NLRP3 and NLRC2 (also named NOD2). NLRP3 does not directly bind the viral RNA. The viral ssRNA or dsRNA causes many intracellular changes (such as reactive oxygen species (ROS) formation and lysosomal maturation), NLRP3 is sensitive to these changes and forms oligomers interacting with ASC (apoptosis‐associated speck‐like protein) and procaspase‐1, collectively called ‘inflammasome complex’. Subsequently, procaspase‐1 is activated, thus leading to the maturation of pro‐IL‐1β and pro‐IL‐18. NLRC2 directly interacts with ssRNA, then it recruits MAVS to activate the IRF3 pathway. Also, it can activate TRAF6 to stimulate the NF‐κB pathway. (B) The JAK‐STAT signaling pathway. IFNα or IFNβ are produced via the IRF3/7 pathway. They bind the IFNAR1/2 (interferon alpha/beta receptor 1/2), leading to the activation of TYK2 (tyrosine kinase 2) and JAK1 (Janus kinase 1). These kinases phosphorylate STAT1 (signal transducer and activator of transcription 1) and STAT2. Subsequently, the phosphorylated STAT1/2 interact with IRF9 to form ISGF3 (IFN‐stimulated gene factor 3). This ternary complex enters the nucleus and promotes the expression of ISGs (interferon‐stimulated genes), such as ISG15, to establish the antiviral status. ISG15 covalently binds target proteins (ISGylation). Coronavirus PL pro can remove ISG15 from ISGylated proteins. Other proteins and abbreviations in this figure: Riplet: an E3 ubiquitin ligase and upstream regulator of RIG‐I; MFN 1/2: mitofusins 1/2. MFN1 and MFN2 regulate mitochondrial fusion; MFN1 is required for the RLR signaling pathway; IMPβ1: importin β1, a nucleocytoplasmic transport receptor, plays roles in the nucleocytoplasmic trafficking of IRF3 as well as NF‐κB p65; MDM2: a p53 degradation stimulator blocks the p53–IRF7–IFNβ signaling pathway; ADNP: activity‐dependent neuroprotective protein, a transcription factor, can bind to IFNα promoter sites (IPS) upon induction by Lpro; MC: mitochondrion; ER: endoplasmic‐reticulum.
The various pathways to interferon expression in the host innate immune system
Viral infection will trigger immune responses. The host immune system consists of innate and adaptive immunity. The innate immune system is the first‐line defense to counteract viral invasion. When viruses enter host cells or replicate in them, various pathogen‐associated molecular patterns (PAMPs) of viruses will be detected by the corresponding pattern‐recognition receptors (PRRs) of the host. Subsequently, PRRs stimulate the different innate immune signaling pathways to produce various antiviral cytokines, including type‐I and type‐III IFNs. Type‐I IFNs include multiple IFNα subtypes and IFNβ, which are produced by virtually all cell types. However, plasmacytoid dendritic cells (pDCs) are the dominant producers of type‐I IFNs 28. Type‐III IFNs comprise IFNλ1‐4 and can also be produced by pDC cells 29. Following their production, IFNs stimulate the antiviral response by binding to different receptors located on the surface of host cells. IFNα/β bind to interferon alpha/beta receptors 1 and 2 (IFNAR1 and 2) (Fig. 2) 30, while IFNλ interacts with interferon lambda receptor 1 and the interleukin 10 receptor subunit beta (IFNLR1 and IL10Rβ) 29. Although the receptors are different between type‐I and type‐III IFNs, they both activate the JAK‐STAT (Janus kinase 1‐signal transducer and activator of transcription 1) pathway to establish the antiviral state 29. Thus, when activated as a consequence of the interaction of IFNα/β with IFNAR1/2, JAK1 and TYK2 (tyrosine kinase 2) phosphorylate STAT1 and STAT2. Subsequently, STAT1/2 interact with IRF9 (interferon regulatory factor 9) to form ISGF3 (IFN‐stimulated gene factor 3). This ternary complex enters the nucleus and activates the expression of various ISGs (IFN‐stimulated genes), such as ISG15. Finally, ISGs utilize autocrine and paracrine signaling to establish an antiviral state in surrounding cells.
Three main PRRs are involved in recognizing RNA viruses, namely Toll‐like receptors (TLRs), retinoic acid‐inducible gene‐I (RIG‐I)‐like receptors (RLRs), and nucleotide‐binding oligomerization domain (NOD)‐like receptors (NLRs) 31, 32, 33. Each of them can trigger the immune response by specific signaling pathways (Fig. 2). The former two PRRs are connected to the expression of type‐I and type‐III IFNs, ISGs, and tumor necrosis factors (TNFs), while NLRs mainly affect interleukin‐1β (IL‐1β) and IL‐18 maturation 33, 34, 35, 36.
Toll‐like receptors (TLRs)
TLRs are transmembrane glycoprotein receptors. Different TLRs can be localized to the surface of the cell or to intracellular endosomes as well as lysosomes; therefore, they can detect various pathogens outside and inside of host cells 37, 38. TLRs comprise three subdomains: the N‐terminal PAMP‐binding region (PBR), which includes multiple leucine‐rich repeats (LRRs), the middle transmembrane domain, and the C‐terminal Toll/IL‐1R homology (TIR) domain (Fig. 3) 38. The N‐terminal domain is used to bind PAMPs, while the C‐terminal domain is involved in initiating the signaling cascades. In man, a total of 10 TLRs have been characterized 39. TLR3, 7, 8 detect various RNA viruses and TLR9 detects DNA viruses 37; in particular, TLR3 is mainly responsible for recognizing dsRNA while TLR7 detects ssRNA 33, 37. However, TLR3 and TLR7 utilize different downstream adaptors to activate the IRF3/7 or the NF‐κB signaling pathway.
Figure 3.
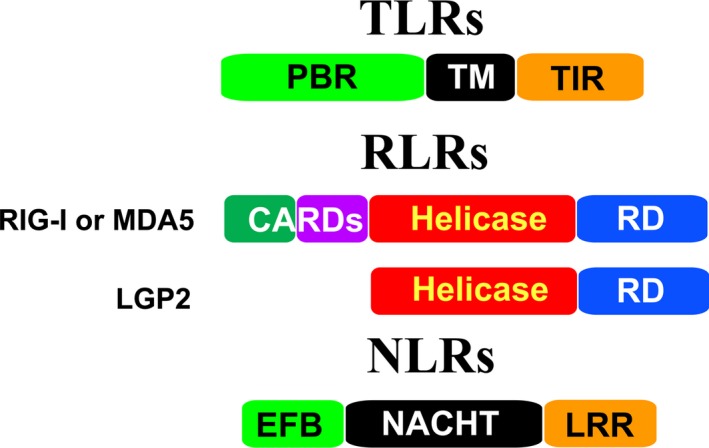
Schematic presentation of individual TLR, RLR, and NLR domains. TLRs contain three domains: the N‐terminal PAMP‐binding region (PBR), the transmembrane region (TM), and the C‐terminal intracellular Toll/IL‐R homology (TIR) domain. RIG‐I and MDA5 comprise an N‐terminal two‐CARDs (caspase‐recruiting domains) domain, the central helicase domain, and the C‐terminal repressor domain (RD). The CARDs domain is absent in LGP2. NLRs have various domain architectures. They mainly contain three domains: the variable N‐terminal effector‐binding domain (EFB), the middle NACHT (domain existing in NAIP,CIITA,HET‐E and TP‐1) domain, and the C‐terminal leucine‐rich repeat (LRR) domain.
The TLR3 signaling pathway
When binding dsRNA, TLR3 dimerizes 38, 40. The C‐terminal TIR domains of dimeric TLR3 interact with the adaptor TRIF (TIR domain‐containing adaptor protein‐inducing IFNβ) 41. TRIF, in turn, recruits Lys63‐linked polyubiquitinated TRAF6 (TNF receptor‐associated factor 6), thereby leading to the Lys63‐linked polyubiquitination of TAK1 (TGF‐β activated kinase 1); TAK1 further recruits TAB 1/2/3 (TAK1‐binding protein 1/2/3), thus yielding the quaternary TAK1/TAB 1/TAB 2/TAB 3 complex, which activates IKKα/β/γ (IκB kinase alpha, beta and gamma; IKKγ is also known as NEMO, NF‐κB essential modulator) 42, 43, 44, 45. Subsequently, the IKKs mediate the phosphorylation and degradation of the NF‐κB inhibitor, IκB. Without IκB binding, NF‐κB will enter the nucleus and trigger the expression of inflammatory genes (Fig. 2A).
Also, TRIF can interact with TRAF3 modified by Lys63‐linked polyubiquitination, in order to further recruit TBK1 (TANK‐binding kinase 1) and IKKε (IκB kinase epsilon) 41, 46. IKKγ (NEMO) is also involved in activating TBK1 and IKKε (Fig. 2A) 47. The activated TBK1/IKKε will phosphorylate IRF3 48, 49, thereby inducing IRF3 dimerization and nuclear translocation to trigger type‐I (mainly IFNβ) and type‐III IFN production (Fig. 2A). Subsequently, the expression of IRF7 is upregulated. TBK1/IKKε also phosphorylates IRF7 49, the activated IRF7 can stimulate the release of various IFNs (Fig. 2A).
The TLR7 signaling pathway
Upon PAMP binding, TLR7 recruits the adaptor MyD88 (myeloid differentiation primary response gene 88) 50. Next, MyD88 forms a complex with interleukin‐1 receptor‐associated kinases (IRAKs) (such as IRAK4, IRAK1, and IRAK2) 37, 51. Subsequently, IRAK4 activates IRAK1/2 and then IRAKs dissociate from upstream MyD88 and interact with downstream TRAF6 51, further activating the NF‐κB signaling pathway as described above (Fig. 2A). Meanwhile, multiple proteins, TRAF6, TRAF3, IRAK4, IRAK1, and IKKα can form a complex for signaling. When contacting this complex, IRF7 is phosphorylated by both IRAK1 and IKKα, thus activating the downstream signaling 37, 51.
RIG‐I‐like receptors (RLRs)
Currently, RLRs comprise three cytosolic proteins: RIG‐I 52, melanoma differentiation‐associated antigen 5 (MDA5) 53, and ‘laboratory of genetics and physiology 2’ (LGP2) 54. RIG‐I and MDA5 each contain two N‐terminal cysteine‐aspartic protease (caspase)‐recruiting subdomains (CARDs), a central DExD/H helicase domain, as well as the C‐terminal repressor domain (RD) (Fig. 3). In contrast, LGP2 lacks the CARDs domain.
The helicase and RD domains of the RLRs are involved in recognizing the dsRNA of RNA viruses 52, while the CARD subdomains lead to intracellular signaling events 31, 52. Due to the absence of CARDs, LGP2 plays a role in regulating RIG‐I and MDA5 signaling. Interestingly, LGP2 is a negative regulator of RIG‐I signaling 54, 55, while it acts a positive regulator facilitating MDA5 binding to viral RNA, thereby augmenting the MDA5 signaling pathway 56, 57. RIG‐I preferentially binds short dsRNA (< 1 kb), while MDA5 binds long dsRNA (> 2 kb) 31. Furthermore, RIG‐I is critical for detecting paramyxoviruses, influenza virus, and Japanese encephalitis virus (JEV), whereas MDA5 recognizes mainly picornaviruses as well as HCV 58, 59.
When RIG‐I binds the PAMPs, the CARDs domain is exposed to interact with the CARD domain of MAVS (mitochondrial antiviral‐signaling protein; also known as IPS‐1, Cardif, VISA), which is localized to the membrane of mitochondria 60. Also, MAVS forms oligomers via its CARD domain, a process that is necessary for downstream signaling 61. At the same time, RIG‐I binding to MAVS is promoted by ubiquitination through TRIM25 62. The RIG‐I‐MAVS interaction stimulates TRAF3 and TRAF6 63, 64. Subsequently, the signals are transduced by the downstream complexes TBK1–IKKε and IKKα/β/γ to further induce the activation of the IRF3 and NF‐κB pathways, respectively, similarly to the downstream signaling pathway of TLR3 mentioned above (Fig. 2A). Finally, these signals trigger the production of IFNs and other host cytokines.
NOD‐like receptors (NLRs)
NOD‐like receptors are cytosolic proteins. They are activated in response to PAMPs in the cytosol (Fig. 2A). The members of the NLR family have various domain architectures 65. However, they contain three common subdomains: (a) the N‐terminal effector‐binding domain (EFB), for example, a CARD domain, a pyrin domain (PYD), or a baculovirus inhibitor of apoptosis protein repeat (BIR) domain; (b) the middle NACHT domain (exists in NAIP, CIITA, HET‐E, and TP‐1); (c) the C‐terminal LRR domain (Fig. 3). According to the different N‐terminal domains, the NLRs are divided into five families: NLRA (N‐terminal acidic activation domain), NLRB (N‐terminal BIR domain), NLRC (N‐terminal CARD domain), NLRP (N‐terminal PYD domain), and NLRX (N‐terminal unknown domain) 65. Here, we discuss two well‐characterized NLRs, NLRP3 and NLRC2, that regulate host immune responses during viral infection.
NLRP3 is sensitive to infection by several (−)ssRNA and (+)ssRNA viruses, such as influenza A virus (IAV), vesicular stomatitis virus (VSV), encephalomyocarditis virus (EMCV), and HCV 66, 67, 68. When NLRP3 is activated by PAMPs derived from invasive viruses, oligomeric NLRP3s interact with ASC (apoptosis‐associated speck‐like protein) and procaspase‐1 (Fig. 2A). They form an inflammasome complex, which results in the activation of caspase‐1, thus leading to the maturation of pro‐IL‐1β and pro‐IL‐18 (Fig. 2A) 36, 66, 67, 68. However, so far, there is no evidence for NLRP3 directly binding virus ssRNA or dsRNA. NLRP3 inflammasome activation relies on lysosomal maturation and the production of reactive oxygen species (ROS) during IAV infection 66; therefore, NLRP3 is likely activated by intracellular changes (such as lysosomal maturation and ROS formation) but not directly by interaction with PAMPs 33, 36.
NLRC2 (alternatively named NOD2), as an intracellular innate immune sensor, recognizes bacterial MDP (muramyl dipeptide) to regulate the host immune response 69. It also recognizes the RNA of several (−)ssRNA viruses, such as IAV, VSV, and respiratory syncytial virus (RSV) 70. NLRC2 directly interacts with viral ssRNA and recruits MAVS to activate the IRF3 pathway, thereby releasing the type‐I IFN, IFNβ 70. NLRC2 can also activate TRAF6 and stimulate the NF‐κB pathway 71.
RNA‐virus proteases interfering with the host innate immune response
Viral proteases are not only important for processing the polyproteins in (+)ssRNA viruses but are also involved in counteracting the host innate immune response. In this review, we focus on the viral proteases of three RNA virus families (Picornaviridae, Coronaviridae, and Flaviviridae) and discuss how they antagonize the host's antiviral response.
The 2A and 3C proteases of Picornaviridae
Two viral proteases, the 2Apro and the 3Cpro, are required for processing the polyprotein in the viral life cycle 4. However, several picornaviruses, such as Foot‐and‐Mouth disease virus (FMDV), have yet another protease, the Lpro 4. Interestingly, hepatitis A virus (HAV) has exclusively the 3C protease 72, whereas the 2A protein is part of the virion 73. Picornaviruses are detected by RLRs, mainly by MDA5, as well as by TLRs 74, 75. It is no surprise that picornaviruses have evolved efficient ways to inhibit the antiviral type‐I IFN production. In particular, the 2Apro and the 3Cpro can disrupt the RLR‐ or TLR‐mediated innate immune pathways. We will discuss further below how the Lpro counteracts the host innate immune response.
2Apro antagonizes the host immune response
The members of the genus Enterovirus of the Picornaviridae family have been well investigated regarding the mechanism of innate immunity disruption by the 2Apro 76, 77, 78. The enteroviral 2Apro possesses a Cys–His–Asp catalytic triad 16. It cleaves between P1 and P2, that is, the site between the capsid and nonstructural‐protein precursors, which is an essential event in the enterovirus life cycle 4. Few inhibitors of the enteroviral 2Apro, let alone an antiviral drug targeting this enzyme, have been described so far. The peptide LVLQTM was shown to antagonize enterovirus A71 (EV‐A71) 2Apro through binding to its active site 79.
2Apro cleaves MDA5 and MAVS
The picornavirus 2Apro modulates the MDA5–MAVS‐mediated antiviral pathway. The Coxsackievirus B3 (CVB3), poliovirus (PV), and EV‐A71 2Apros were shown to cleave both MDA5 and MAVS, leading to inhibition of IFNβ and type‐III IFN (IFNλ1‐4) production (Fig. 2A) 76, 77, 78. Feng et al. 77 concluded that enteroviruses use a common strategy to antagonize the host IFN response. Furthermore, these authors found that MDA5 and MAVS were degraded by a caspase‐proteasome‐independent pathway in CVB3‐infected cells. In contrast, Barral et al. 80 reported that MDA5 was cleaved via the caspase‐proteasome‐dependent pathway in PV infection.
2Apro cleaves TRIF
TRIF is an important adaptor in the TLR3 pathway 41. The protein level of TRIF is reduced in CVB3‐infected cells due to the 3Cpro 81 (see below). In 2016, Lind et al. 78 found that CVB3 2Apro also cleaves TRIF (Fig. 2A), and further antagonizes type‐I and type‐III IFN production.
Does the 2Apro affect the JAK‐STAT pathway?
EV‐A71 infection leads to increased IFNβ levels but inhibits the transcription of ISGs in vivo 82. Lu et al. 82 found that the 2Apro reduces IFNAR1 expression levels to impede the JAK‐STAT pathway (Fig. 2B), thereby leading to a decreased production of ISGs. Furthermore, the protease activity of the 2Apro was essential for downregulating IFNAR1. However, Liu et al. 83 reported that EV‐A71 infection did not alter IFNAR1 but instead JAK1 expression in vivo. Furthermore, overexpressing viral 2Apro (or 3Cpro) did not affect the JAK1 expression level. Conclusively, these authors demonstrated that the 2Apro does not act as an antagonist to the JAK‐STAT pathway, although EV‐A71 infection does affect this signaling pathway by inhibiting JAK1 expression 83. Very recently, Wang et al. reported that EV‐A71 infection leads to degradation of karyopherin‐α1 (KPNA1), a nuclear localization signal receptor for phosphorylated STAT1. Thus, STAT1 transport into the nucleus is blocked, thereby shutting off the JAK‐STAT pathway 84. Interestingly, these authors found that neither the 2Apro nor the 3Cpro is the culprit here; instead, it is caspase‐3 activated by the virus infection that degrades KPNA1 84. However, it is still unclear whether 2Apro can affect the JAK‐STAT pathway by other mechanisms or does not affect this signaling pathway at all.
Finally, the 2Apro can degrade PABP (poly(A)‐binding protein) and eIF4G (eukaryotic initiation factor 4G) to shut down the host translation machinery 85, 86, 87, thereby globally inhibiting the production of antiviral host proteins.
3Cpro antagonizes the host immune response
The 3Cpro has either a catalytic Cys–His dyad or a Cys–His–Asp/Glu triad in different picornaviruses (Fig. 1B) 17, 18, 19. The protease prefers to cleave between Gln and Gly (and sometimes, between Glu and Gly) 88. Besides viral polyprotein processing, this protease also has an RNA‐binding activity being essential for viral RNA replication 89. Currently, Michael‐acceptor compounds such as rupintrivir and SG85 have been described as potent, broad‐spectrum inhibitors of enterovirus 3C proteases 19, 90, 91.
3Cpro modulates RIG‐I, MDA5, and MAVS
The picornavirus 3Cpro inhibits the RLR signaling pathway. EV‐A71 3Cpro has been reported to bind the N‐terminal CARDs of RIG‐I without digesting RIG‐I, thereby inhibiting the recruitment of MAVS and disrupting the type‐I IFN response 92. At variance with this report, Feng et al. 77 found that not only the EV‐A71 enzyme but also the CVB3 and PV 3Cpros do cleave RIG‐I in vivo. EMCV 3Cpro can cleave RIG‐I in vitro 93. A caspase‐mediated degradation of RIG‐I was also observed in EMCV‐infected cells 93. The exact mechanism of the RIG‐I regulation by 3Cpro needs to be further investigated; in any case, the partly conflicting observations mentioned above indicate that RIG‐I is an important target antagonized by the picornavirus 3Cpro.
Like RIG‐I, MDA5 binds MAVS to activate downstream cascades of the innate immune system (see above). Lei et al. 92 found by co‐immunoprecipitation (co‐IP) that EV‐A71 3Cpro can bind MDA5 (Fig. 2A). Furthermore, Rui et al. 94 detected by co‐IP that the CV‐A16, CV‐A6, or EV‐D68 3Cpros can also bind MDA5, thus disrupting the MDA5–MAVS interaction. Interestingly, these 3Cpros do not digest MDA5 and a proteolytically inactive, mutated 3Cpro can also prevent MDA5 from activating IFN.
Differently, HAV can cleave MAVS through the 3ABC precursor protein [the 3A (a membrane anchor protein), 3B (VPg) plus protease domain] but not through the mature 3Cpro alone 95. Mukherjee et al. 81 demonstrated that MAVS was cleaved between Gln148 and Ala149 by the 3Cpro in CVB3‐infected cells. Recently, the picornavirus Seneca Valley virus (SVV) 3Cpro was also shown to induce cleavage of MAVS at the same position, Gln148↓Ala149 (↓: cleavage site) 96. Therefore, both viruses suppress the antiviral IFN production through cleavage of MAVS.
3Cpro cleaves TRIF
The picornavirus 3Cpro can also interfere with the TLR3 pathway. As mentioned above, the C‐terminal TIR domains of dimeric TLR3 interact with TRIF to stimulate the downstream IRF3 and NF‐κB activities. CVB3 3Cpro was demonstrated to cleave TRIF (Fig. 2A), thereby blocking the downstream type‐I IFN production in CVB3‐infected cells 81. In total, five cleavage sites in TRIF (Gln190↓Gly191, Gln653↓Ser654, Gln659↓Ser660, Gln672↓Ser673, and Gln702↓Ala703) were found 81. Furthermore, the 3Cpro of EV‐A71 or EV‐D68 can also cleave TRIF, leading to inactivation of the signaling along the IRF3 and NF‐κB pathways in vivo 97, 98. However, the EV‐A71 3Cpro processes only one site between Gln312 and Ser313, while the EV‐D68 protease cleaves two sites (Gln312↓Ser313, Gln653↓Ser654) 97, 98. It seems that the different 3Cpros possess slightly different cleavage patterns on TRIF. Whether these subtleties are linked to any difference in biological response needs to be answered. Similarly to the 3ABC precursor digesting MAVS (see above), the HAV protease can also cleave TRIF in vivo 99, but the degradation appears to be exclusively performed by the 3CD (protease‐polymerase precursor) protein, not by mature 3C protease alone. The two cleavage sites in TRIF are Gln190↓Gly191 and Gln554↓His555 99. The observation that some precursor proteins (such as HAV 3ABC, 3CD) seem to be involved in counteracting the host immune response should motivate further investigations into more protease precursors, instead of looking only into mature proteases.
3Cpro cleaves IKKγ (also named NEMO)
The FMDV 3Cpro can process the porcine IKKγ (also called NEMO) at the unusual cleavage site Gln384↓Arg385, thereby removing the C‐terminal zinc‐finger domain of this protein 100 (these authors erroneously misnumbered Gln384 as Gln383), thus blocking the signaling pathways of NF‐κB and IRF3. From the same group, Wang et al. 101 reported that mature HAV 3Cpro can cleave NEMO at position Gln304↓Ala305, thereby antagonizing type‐I IFN production. These authors also reported that the precursor proteins 3ABC or 3CD of HAV can also cleave NEMO but with less efficiency 101.
3Cpro cleaves TANK
TANK (TRAF family member‐associated NF‐κB activator), as a positive regulator, interacts with the TBK1/IKKε complex to enhance type‐I IFN production 102. However, the role of TANK in regulating the NF‐κB pathway is a matter of debate. Chariot et al. 103 reported that TANK binds IKKγ (NEMO) and upregulates the NF‐κB pathway. Blocking the TANK–IKKγ interaction by deleting the TANK‐binding domain of IKKγ impairs NF‐κB activation 103. However, Lys63‐linked polyubiquitination of TRAF6 is required for NF‐κB activation 104. Wang et al. 105 proposed that TANK acts as a negative regulator of the NF‐κB pathway by inducing TRAF6 deubiquitination.
The EMCV 3Cpro can cleave TANK at two sites, Gln197↓Ala198 and Gln291↓Gly292 106. Huang et al. 106 reported that NF‐κB signaling increased when TANK was processed by EMCV 3Cpro. According to Wang et al.'s proposal 105 (see above), the explanation might be that the degradation of TANK can release TANK–TRAF6‐mediated NF‐κB inhibition. From the same group, Huang et al. 107 reported that the EMCV 3Cpro can disrupt the TANK–TBK1–IKKε–IRF3 complex by cleaving TANK, thus decreasing type‐I IFN production. It is of interest to further investigate the reason why the EMCV 3Cpro processes TANK to stimulate the NF‐κB pathway but downregulate the IRF3 pathway. Very recently, Qian et al. 96 showed that the SVV 3Cpro processes TANK at two cleavage sites, Glu272↓Phe273 and Gln291↓Gly292. Whereas the former site is unusual because of the large P1' residue, the latter is also cleaved by the EMCV 3Cpro, indicating that it may be a conserved target on TANK for different picornavirus 3Cpros.
3Cpro cleaves TAK1/TAB 1/TAB 2/TAB 3
The EV‐A71 3Cpro uses another mechanism to deactivate the NF‐κB pathway. As mentioned above, TAK1 can bind TAB 1 to form a complex 42. This complex recruits TAB 2 and TAB 3, yielding the quaternary TAK1/TAB 1/TAB 2/TAB 3 43, 44. The combination of TAK1 and TABs can activate IKKs (such as IKKα, IKKβ, and IKKγ) 45, thereby upregulating the NF‐κB pathway. The EV‐A71 3Cpro cleaves TAK1 (Gln360↓Ser361), TAB 1 (Gln414↓Gly415 and Gln451↓Ser452), TAB 2 (Gln113↓Ser114), and TAB 3 (Gln173↓Gly174 and Gln343↓Gly344) in vivo (Fig. 2A) 108. Recently, the CV‐A16, CV‐A6, and EV‐D68 3Cpros have also been shown to process TAK1 94. In summary, the enterovirus 3Cpro impairs NF‐κB activation.
3Cpro cleaves IRFs
IRF7 stimulates type‐I IFN production, such as IFNα, thereby activating the JAK‐STAT pathway in adjacent cells 109. The 3Cpro of EV‐A71 can process IRF7 at a cleavage site between Gln189 and Ser190 in vitro and in vivo, while the 3Cpro of EV‐D68 does so at two sites (Gln167↓Ala168 and Gln180↓S190) 110, 111.
Furthermore, IRF9 interacts with STAT1 and STAT2 to form a complex, ISGF3 in the JAK‐STAT pathway, thereby stimulating ISG production (Fig. 2B) 30, 112. The EV‐A71 3Cpro can cleave IRF9 in EV‐A71‐infected cells as well as in an in vitro assay, resulting in reduced IFN signaling 113.
Finally, the 3Cpro can digest PABP to shut off host translation 114, 115, thereby globally inhibiting the production of antiviral proteins.
Lpro interferes with the host innate immune response
So far, the roles of the Lpro have been well investigated for FMDV. Due to initiation at different AUG codons, two forms of the Lpro, Labpro and Lbpro, were discovered in FMDV 116. The FMDV Lpro, a papain‐like protease, contains a Cys–His catalytic dyad 25.
The FMDV Lpro can degrade p65/RelA, a subunit of NF‐κB, to block the NF‐κB activity 117. de los Santos et al. 118 found that a putative SAP (SAF‐ACINUS‐PIAS) domain of the Lpro affects its subcellular localization, thus further mediating the degradation of p65/RelA. Also, the FMDV Lpro can decrease the IRF3 and IFR7 protein levels in vivo 119, but the corresponding mRNAs are apparently not affected.
Medina et al. 120 recently reported that the FMDV Lpro binds the host transcription factor ADNP (activity‐dependent neuroprotective protein) in vitro and in vivo. In addition, these authors found that wild‐type FMDV but not ΔLpro FMDV can induce ADNP to bind to IFNα promoter sites (IPS; Fig. 2A), thus disrupting the expression of IFN and ISGs 120.
Furthermore, the FMDV Lbpro exhibits deubiquitinating activity 121; it can remove Lys48‐ and Lys63‐linked polyubiquitin in vitro and in vivo. As mentioned above, the ubiquitination of several elements is essential in innate immune pathways, for example, of RIG‐I, TRAF3, and TRAF6 62, 104, 122. Wang et al. 121 reported that the FMDV Lbpro can deubiquitinate RIG‐I, TBK1, TRAF3, and TRAF6 in HEK293T cells overexpressing this protease, which leads to blocking type‐I IFN production.
Like the 2Apro, the Lpro can also cleave eIF4G to globally inhibit the translation of mRNAs coding for host antiviral proteins 25, 87.
Many investigations have shown that the papain‐like proteases of coronaviruses also possess DUB activity relevant for antagonizing the host innate immune response; these enzymes are discussed in the next paragraph.
Papain‐like proteases and 3C‐like proteases of the Coronaviridae interfere with innate immunity
Belonging to the family Coronaviridae, coronaviruses have one or two viral PLpro(s) and one 3CLpro (Mpro) 9. The PLpro(s) is (are) part of the nonstructural protein 3 (Nsp3) 123, 124. PLpro is a cysteine protease with a catalytic triad Cys–His–Asp (Fig. 1B) 26. The CoV PLpro has proteolytic, deubiquitinating, and deISGylating (removal of ISG15 from target proteins) activities 26, 27, 125; through the latter two, it can disrupt the host immune response. Thus far, only a few inhibitors targeting the PLpro have been described. Báez‐Santos et al. 126 demonstrated that several naphthalene derivatives efficiently block the enzymatic activity of SARS‐CoV PLpro.
PLpro modulates the TLR7 pathway by deubiquitinating TRAF3 and TRAF6
As mentioned above, TLR7 recognizes ssRNA 33, 37. In 2013, Li et al. 127 showed that GU‐rich ssRNA of SARS‐CoV can be detected by TLR7.
The SARS‐CoV PLpro removes Lys63‐linked ubiquitin (Ub) chains from TRAF3 and TRAF6 to reduce the TLR7‐mediated immune signaling 128. Furthermore, the SARS‐CoV PLpro cannot remove Lys48‐linked Ub chains from TRAF3 and TRAF6 according to a western blot assay 128. This is at variance with an in vitro study that demonstrated that SARS‐CoV PLpro prefers to process Lys48‐ over Lys63‐linked polyUb chains 129. Perhaps, one might speculate that the target proteins, TRAF3 or TRAF6, affect the PLpro DUB specificity.
In addition, TRAF3 and TRAF6 are two key components of the RLR pathways (Fig. 2A); therefore, we have reason to believe that PLpro could also regulate RLR signaling by deubiquitinating these two targets.
PLpro modulates the STING–TRAF3–TBK1–IKKε complex
STING (stimulator of IFN genes, also known as MITA, ERIS) is a protein of the endoplasmic‐reticulum (ER) (Fig. 2A). It can activate the IRF3 pathway upon dimerization and ubiquitination as well as upon interaction with several partners, such as MAVS, TRAF3, TBK1, and IKKε 130, 131. The SARS‐CoV PLpro+TM (TM: transmembrane region of Nsp3; see Ref. 124 for a review) and Human Coronavirus‐NL63 (HCoV‐NL63) PL2pro+TM bind STING in a co‐IP assay 132. These two proteins inhibit the dimerization and ubiquitination of STING and disrupt STING interaction with other partners, thereby blocking STING‐mediated IFN production. Interestingly, the enzymatic activity of SARS‐CoV PLpro+TM or HCoV‐NL63 PL2pro+TM is not required for modulating STING 132.
Furthermore, SARS‐CoV PLpro+TM can disrupt the STING–TRAF3–TBK1–IKKε complex by binding each of its components 133. In addition, the SARS‐CoV PLpro+TM or PLpro alone were also reported to bind IRF3 133, 134. Meanwhile, SARS‐CoV PLpro reduces the ubiquitination of STING, TRAF3, and TBK1 133. Also, murine hepatitis virus A59 (MHV‐A59) PL2pro deubiquitinates TBK1 and binds TBK1 and IRF3 135, 136. All these observations demonstrate that the PLpro is heavily involved in regulating the IRF3 pathway.
PLpro blocks the p53–IRF7–IFNβ pathway
The tumor suppressor protein p53 enhances the antiviral type‐I IFN response 137. In 2015, Yuan et al. 138 found that p53 can upregulate the transcription of IRF7. HCoV‐NL63 PL2pro deubiquitinates and stabilizes MDM2, a p53 degradation stimulator, thus causing p53 degradation and blocking the p53–IRF7–IFNβ signaling pathway (Fig. 2A) 138. p53 also inhibits SARS‐CoV replication 139. Ma‐Lauer et al. 139 found that the PLpros of SARS‐CoV and MERS‐CoV as well as the two PLpros of HCoV NL63 can bind RCHY1, another p53‐degradation stimulator. The SARS‐CoV ‘unique domain' (SUD) enhances the interaction between the PLpro and RCHY1. This interaction increases the stability of RCHY1, thereby stimulating p53 degradation 139. In conclusion, the coronavirus PLpro utilizes various ways to modulate p53 and further regulate host innate immunity responses.
PLpro blocks the NF‐κB pathway
Besides blocking the IRF3 pathway, Frieman et al. 140 reported that the SARS‐CoV PLpro stabilizes IκBα, an inhibitor of NF‐κB, to modulate the NF‐κB signaling pathway. Further, these workers found that the HCoV‐NL63 but not the MHV PL2pro can counteract the IRF3 and NF‐κB pathways 140. These observations indicate that the functions of the PLpro could be specific for different CoVs.
Furthermore, Devaraj et al. 134 indicated that the protease activity of the SARS‐CoV PLpro is not required for blocking IFNβ production. Frieman et al. 140 reported that the enzyme activity of SARS‐CoV PLpro is dispensable for IFNβ production via the IRF3 pathway but not for TNFα production through the NF‐κB pathway. In 2010, Clementz et al. 141 also found that the catalytic activity of the HCoV‐NL63 PL2pro is not responsible for blocking IFNβ production. In contrast, the MHV PL2pro requires the enzymatic activity for blocking IFNβ induction 136. The exact relationship between the enzymatic activity of the PLpro and IFN production is still a matter of debate today.
Other roles of the PLpro in counteracting host immunity
The PLpro has deISG15ylating activity (Fig. 2B) 125, leading to the downregulation of the host immune response. However, the detailed mechanism of this process is still not completely clear. In addition, autophagy could play a negative role in the host innate immune response 142. Chen et al. 143 found that the PLpro+TM of SARS‐CoV induces incomplete autophagy by interacting with LC3 and Beclin1 (two key autophagy regulators), thus negatively regulating antiviral immunity. Knockdown of Beclin1 could partially reverse the effect of the PLpro on innate immune signaling 143. Therefore, these authors assume that the PLpro+TM inducing autophagy could represent a new mechanism of antagonism to host innate immunity by coronaviruses.
3CLpro antagonizes host immune responses
The 3CLpro (Mpro), the other protease of coronaviruses, is also involved in counteracting the host innate immune response. A number of peptidic and peptidomimetic inhibitors carrying various warheads have been described to block the activity of the coronavirus Mpro (144; see Refs 9, 145 for reviews).
The Mpros of two CoVs infecting pigs have recently been reported to antagonize the host immune response (Fig. 2) 146, 147, 148. The Mpros of both porcine epidemic diarrhea virus (PEDV, a member of the genus Alphacoronavirus) and porcine deltacoronavirus (PDCoV) cleave porcine IKKγ (NEMO) at the identical site, Gln231↓Val232 146, 147. As mentioned above, NEMO is required for activating the NF‐κB and IRF3 pathways. Therefore, the cleavage of IKKγ by PEDV and PDCoV Mpros abrogates NF‐κB signaling and inhibits IFNβ induction 146, 147. In addition, the Mpro of PDCoV can process porcine STAT2 at two sites, Gln685↓Glu686 and Gln758↓Ser759, to impair the JAK‐STAT pathway 148, thereby reducing ISG production. It is interesting to investigate whether the human CoV Mpro exhibits similar activities to affect the host innate immune response.
Proteases of Flaviviridae interfering with the innate immune response
In the following paragraphs, we will discuss the proteases of the genera Hepacivirus, Flavivirus, and Pestivirus of the Flaviviridae family.
HCV NS3/NS4A protease counteracts host innate immune pathways
The best known member of the genus Hepacivirus is HCV. HCV produces two proteases, NS2pro and NS3/NS4Apro 10, 11. The autoprotease NS2pro operates only on one cleavage site, between NS2 and NS3, while the other cleavage sites among the NS proteins of HCV are processed by the NS3/NS4Apro 11. Currently, the NS3/NS4Apro, but not the NS2pro, is reported to be related to counteracting host innate immune responses (Fig. 2A). The former enzyme features the catalytic triad Ser–His–Asp 20. The cleavage site specificity of the NS3/NS4Apro favors Cys or Thr in the P1 position, an acidic residue in the P6 position, and a residue with a small side‐chain (Ala or Ser) in P1′, that is, D/E‐XXXX‐C/T‐S/A for the P6‐P1′ sequence 11. NS4A is a cofactor for the NS3 protease. Several synthetic inhibitors of the HCV NS3/NS4Apro, such as simeprevir and paritaprevir (ABT‐450), have helped to dramatically improve the therapy of liver disease caused by HCV 149, 150.
HCV NS3/NS4Apro cleaves MAVS
In 2005, RIG‐I was shown to detect the 3′ UTR (untranslated region) of the HCV genome 151. However, later Cao et al. 59 demonstrated that MDA5 plays a major role in recognizing the 3′ UTR of HCV while RIG‐I seems to be less important. Subsequently, MDA5 (or RIG‐I) binds MAVS, thereby activating the downstream IRF3 and NF‐κB pathways. HCV NS3/NS4Apro cleaves MAVS (also named Cardif, as mentioned above; Fig. 2A) at the cleavage site Cys508↓His509 152, thereby disrupting the host immune response. The NS3/NS4Apro of GB virus B (GBV‐B), from the same genus as HCV, cleaves MAVS at the same site as the HCV enzyme 153. These authors further found that MAVS was released from the mitochondrial membrane to the cytosol due to this cleavage 153. Because the location of MAVS on the mitochondrial membrane is essential for its functions, this observation could explain how MAVS fails to transmit the signal downstream after being processed by NS3/NS4Apro 153. Furthermore, NS3/NS4A proteases from Hepaciviruses infecting other animals (such as monkeys, rodents, horses, and cows) can cleave their cognate MAVS proteins 154. The cleavage of MAVS presents a common mechanism used by Hepaciviruses to regulate the host immune response.
NS3/NS4Apro cleaves Riplet (upstream regulator of RIG‐I)
As mentioned above, RIG‐I is ubiquitinated by TRIM25 for its activation 62. Oshiumi et al. 155 reported that the protein Riplet (an E3 ubiquitin ligase) is a prerequisite for TRIM25 stimulation of RIG‐I signaling. Knocking out Riplet abrogates the expression of type‐I IFN in response to HCV RNA 155. HCV NS3/NS4Apro can cleave Riplet at position Cys21↓Ile22 in vitro, because the residues 16‐EDDLGC‐21 of Riplet are similar to the consensus cleavage motif D/E‐XXXX‐C/T of HCV NS3/NS4A protease. As a consequence, RIG‐I activation is abolished 155.
NS3/NS4Apro modulates TRIF of the TLR3 pathway
TLR3 is sensitive to the intermediate dsRNA of HCV replication 156. Upon sensitization, its C‐terminal TIR domain can interact with TRIF to stimulate the downstream cascades. HCV NS3/NS4Apro cleaves TRIF at the site Cys372↓Ser373 in vitro and in vivo (HEK293 cells) to disrupt the TLR3 pathway 157. In contrast to this observation, Dansako et al. 158 found that HCV NS3/NS4A cannot cleave TRIF in PH5CH8 (immortalized human hepatocytes), HeLa, and Huh‐7‐derived cells. The effects of the NS3/NS4A protease on TRIF need to be further investigated, in order to resolve this ambiguity.
NS3/NS4Apro modulates importin β1
Very recently, Gagné et al. 159 found that the HCV NS3/NS4Apro can cleave importin β1 (IMPβ1; Fig. 2A). IMPβ1, a nucleocytoplasmic transport receptor, transports proteins from the cytoplasm to the nucleus. The HCV NS3/NS4A protease triggers the degradation of IMPβ1 and inhibits or disrupts the nucleocytoplasmic trafficking of IRF3 as well as NF‐κB p65, thus preventing the host immune response 159.
Flavivirus NS2B/NS3 proteases counteracting host innate immune pathways
Flaviviruses only have one protease, the NS2B/NS3pro 12. This enzyme comprises the N‐terminal third of NS3 and the middle hydrophilic part of NS2B as a cofactor, together forming the NS2B/NS3pro. Like HCV NS3/NS4Apro, the flavivirus NS2B/NS3pro is a chymotrypsin‐like serine protease with a catalytic Ser–His–Asp triad (Fig. 1B) 21, 22. The P1 and P2 positions at the NS2B/NS3pro cleavage sites are conserved as basic residues, Lys or Arg. The NS2B/NS3pro is an attractive antiviral target. This protease is also involved in counteracting host innate immune pathways (Fig. 2A). Currently, no approved drug is available that targets the flavivirus NS2B/NS3pro. Several peptide aldehydes and peptide boronic‐acid inhibitors have been described to inhibit the activity of the flavivirus NS2B/NS3pro 22, 160, 161, 162.
The NS2B/NS3pro cleaves STING
As mentioned above, STING can upregulate IRF3 signaling (Fig. 2A) 130, 131. DENV NS2B/NS3pro cleaves human STING at the site Arg94‐Arg95↓Gly96 in vivo and thereby inhibits induction of IFNβ 163, 164. However, this protease is unable to cleave RIG‐I, TLR3, TBK1, IKKε, IRF3, and IRF7 163. Recently, Liu et al. 165 reported that the full‐length DENV NS3 (including the C‐terminal helicase), but not the NS2B, can be modified by Lys27‐linked polyubiquitination when co‐transfecting Ub and NS3 in HEK 293T cells. The ubiquitinated NS3 facilitates recruitment of NS2B, that is, the formation of the NS2B/NS3 protease, thereby enhancing the cleavage of STING. Furthermore, these authors found that the ER protein SCAP (sterol regulatory element‐binding protein (SREBP) cleavage‐activating protein) can bind NS2B and inhibit polyubiquitination of NS3, thus disrupting the formation of NS2B/NS3pro and the cleavage of STING 165.
The NS2B/NS3 protease cleaves two mitofusins (MFN1, MFN2)
MFN1 and MFN2 regulate the mitochondrial fusion; in particular, MFN1 is required for the RLR signaling pathway 166. Yu et al. 167 demonstrated that the DENV NS2B/NS3 protease can cleave MFN1 (Arg539‐Asn540↓Ala541) and MFN2 (Arg563‐Arg564↓Ala565) in cells. Subsequently, the cleaved MFNs suppress mitochondrial fusion and disrupt IFN production. Interestingly, the homologous protease from JEV cannot cleave MFNs for unknown reasons 167. It is worth investigating whether or not other flavivirus proteases modulate MFNs.
Pestivirus N‐terminal protease counteracts host immune responses
As mentioned above, a total of three proteases – Npro, NS2pro, and NS3/NS4Apro – are encoded by the pestivirus genome. To our knowledge, only the Npro is reported to be involved in counteracting the host innate immune pathways (Fig. 2A); therefore, we will restrict ourselves to discussing this enzyme here. The pestivirus Npro is cleaved off the polyprotein by autolysis between C (the capsid protein) and the Npro 14. It is a cysteine protease with a catalytic dyad Cys–His. The Npro adopts neither a chymotrypsin‐like nor a papain‐like fold. Instead, it features a unique ‘clam‐like’ fold containing a catalytic protease domain and a zinc‐binding domain 168. After autocleavage, the Npro loses its cleavage capability by intramolecular auto‐inhibition 168.
Npro binds IRF3 and IRF7
The Npros of CSFV and BVDV directly bind IRF3 and induce degradation of the factor by the host proteasome, thus interfering with IFN production 169, 170, 171, 172. According to point mutation experiments, both the protease domain and the zinc‐binding domain are essential for Npro binding to IRF3 168, 173, 174.
Furthermore, the CSFV Npro can also bind IRF7 and downregulate the IRF7 protein level in porcine DC cells, thus limiting type‐I IFN production 175. Also, Fiebach et al. 175 found that the zinc‐binding domain but not the protease domain is required to bind IRF7.
Npro interacts with IκBα (NF‐κB inhibitor)
The CSFV Npro binds IκBα in vitro and in vivo 176. However, the enzyme does not affect the NF‐κB activity 176. The BVDV Npro cannot block the NF‐κB pathway either 171. The role of the Npro‐IκBα interaction, thus, remains unclear for the time being.
Conclusions
The proteases of emerging or re‐emerging (+)ssRNA viruses are always worth investigating, either as targets for direct antivirals disrupting polyprotein processing 9, 162, 177 or as important players in mounting the viral anti‐IFN activity. In this review, we discussed the roles of viral proteases from the families Picornaviridae, Coronaviridae, and Flaviviridae in counteracting host innate immune responses. Among the different PRR‐mediated signaling pathways, a large body of data is available for components of the RLR and TLR pathways being cleaved by viral proteases, but much less so for the NLR pathway (with the notable exception of MAVS in the NLRC2 pathway as a prominent target of viral proteases). A possible reason for this could be that at this time, many immunity‐related functional roles of NLRs remain unclear. Another observation is that a multitude of reports exist on the proteolytic cleavage of components of the innate immune system by the picornaviral 2Apro and 3Cpro as well as the HCV NS3/NS4Apro, while proteolytic cleavage by the coronavirus proteases PLpro and 3CLpro (Mpro) seems to be comparatively rare. This may be due to more research having been performed on the picornaviruses and on HCV, or to the fact that the DUB activity of the coronavirus PLpro (as opposed to its proteolytic activity) is a very efficient player in counteracting the innate immune response. Furthermore, it should be remembered that CoVs feature many other proteins (such as ORF3b, ORF6, the nucleocapsid protein, the membrane protein, and the X domain in SARS‐CoV; ORF4a, ORF4b, ORF5, and the membrane protein in MERS‐CoV) that are involved in suppressing IFN production 124, 178, so proteolytic cleavage of host immunity proteins is perhaps required to a lesser extent here. For picornaviruses and HCV on the other hand, there is only occasional reports on nonproteases, such as the picornaviral 2C and 3A proteins 179, 180 as well as HCV NS5A and E2 181, 182, being involved in diminishing cytokine production.
In general, homologous proteases from the same family show common mechanisms in regulating host immunity pathways. For example, all 3Cpros from CVB3, EV‐A71, and SSV cleave TRIF. However, occasionally homologous enzymes exhibit some unique characteristics; thus, the DENV but not the JEV NS2B/NS3 protease can cleave MFNs. All these findings not only contribute to our understanding of the host's immune response to viral infection but can also help us discover broad‐spectrum or specific antiviral drugs targeting viral proteases and their interaction with host signaling pathways.
Edited by Wilhelm Just
References
- 1. Adams MJ, Lefkowitz EJ, King AMQ, Harrach B, Harrison RL, Knowles NJ, Kropinski AM, Krupovic M, Kuhn JH, Mushegian AR et al (2017) Changes to taxonomy and the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses. Arch Virol 162, 2505–2538. [DOI] [PubMed] [Google Scholar]
- 2. Jiang P, Liu Y, Ma HC, Paul AV and Wimmer E (2014) Picornavirus morphogenesis. Microbiol Mol Biol Rev 78, 418–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Norder H, De Palma AM, Selisko B, Costenaro L, Papageorgiou N, Arnan C, Coutard B, Lantez V, De Lamballerie X, Baronti C et al (2011) Picornavirus non‐structural proteins as targets for new anti‐virals with broad activity. Antiviral Res 89, 204–218. [DOI] [PubMed] [Google Scholar]
- 4. Seipelt J, Guarné A, Bergmann E, James M, Sommergruber W, Fita I and Skern T (1999) The structures of picornaviral proteinases. Virus Res 62, 159–168. [DOI] [PubMed] [Google Scholar]
- 5. Eckerle I, Müller MA, Kallies S, Gotthardt DN and Drosten C (2013) In‐vitro renal epithelial cell infection reveals a viral kidney tropism as a potential mechanism for acute renal failure during Middle East Respiratory Syndrome (MERS) Coronavirus infection. Virol J 10, 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brierley I, Digard P and Inglis SC (1989) Characterization of an efficient coronavirus ribosomal frameshifting signal: requirement for an RNA pseudoknot. Cell 57, 537–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Neuman BW and Buchmeier MJ (2016) Supramolecular architecture of the coronavirus particle. Adv Virus Res 96, 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu DX, Fung TS, Chong KK, Shukla A and Hilgenfeld R (2014) Accessory proteins of SARS‐CoV and other coronaviruses. Antiviral Res 109, 97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hilgenfeld R (2014) From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J 281, 4085–4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Grakoui A, McCourt DW, Wychowski C, Feinstone SM and Rice CM (1993) A second hepatitis C virus‐encoded proteinase. Proc Natl Acad Sci USA 90, 10583–10587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bartenschlager R, Ahlborn‐Laake L, Yasargil K, Mous J and Jacobsen H (1995) Substrate determinants for cleavage in cis and in trans by the hepatitis C virus NS3 proteinase. J Virol 69, 198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Falgout B, Pethel M, Zhang YM and Lai CJ (1991) Both nonstructural proteins NS2B and NS3 are required for the proteolytic processing of dengue virus nonstructural proteins. J Virol 65, 2467–2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rümenapf T, Stark R, Heimann M and Thiel HJ (1998) N‐terminal protease of pestiviruses: identification of putative catalytic residues by site‐directed mutagenesis. J Virol 72, 2544–2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lackner T, Müller A, Pankraz A, Becher P, Thiel HJ, Gorbalenya AE and Tautz N (2004) Temporal modulation of an autoprotease is crucial for replication and pathogenicity of an RNA virus. J Virol 78, 10765–10775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tautz N, Elbers K, Stoll D, Meyers G and Thiel HJ (1997) Serine protease of pestiviruses: determination of cleavage sites. J Virol 71, 5415–5422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Petersen JF, Cherney MM, Liebig HD, Skern T, Kuechler E and James MN (1999) The structure of the 2A proteinase from a common cold virus: a proteinase responsible for the shut‐off of host‐cell protein synthesis. EMBO J 18, 5463–5475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Matthews DA, Smith WW, Ferre RA, Condon B, Budahazi G, Sisson W, Villafranca JE, Janson CA, McElroy HE, Gribskov CL et al (1994) Structure of human rhinovirus 3C protease reveals a trypsin‐like polypeptide fold, RNA‐binding site, and means for cleaving precursor polyprotein. Cell 77, 761–771. [DOI] [PubMed] [Google Scholar]
- 18. Wang J, Fan T, Yao X, Wu Z, Guo L, Lei X, Wang J, Wang M, Jin Q and Cui S (2011) Crystal structures of enterovirus 71 3C protease complexed with rupintrivir reveal the roles of catalytically important residues. J Virol 85, 10021–10030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tan J, George S, Kusov Y, Perbandt M, Anemüller S, Mesters JR, Norder H, Coutard B, Lacroix C, Leyssen P et al (2013) 3C protease of enterovirus 68: structure‐based design of Michael acceptor inhibitors and their broad‐spectrum antiviral effects against picornaviruses. J Virol 87, 4339–4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim JL, Morgenstern KA, Lin C, Fox T, Dwyer MD, Landro JA, Chambers SP, Markland W, Lepre CA, O'Malley ET et al (1996) Crystal structure of the hepatitis C virus NS3 protease domain complexed with a synthetic NS4A cofactor peptide. Cell 87, 343–355. [DOI] [PubMed] [Google Scholar]
- 21. Erbel P, Schiering N, D'Arcy A, Renatus M, Kroemer M, Lim SP, Yin Z, Keller TH, Vasudevan SG and Hommel U (2006) Structural basis for the activation of flaviviral NS3 proteases from dengue and West Nile virus. Nat Struct Mol Biol 13, 372–373. [DOI] [PubMed] [Google Scholar]
- 22. Lei J, Hansen G, Nitsche C, Klein CD, Zhang L and Hilgenfeld R (2016) Crystal structure of Zika virus NS2B‐NS3 protease in complex with a boronate inhibitor. Science 353, 503–505. [DOI] [PubMed] [Google Scholar]
- 23. Dubrau D, Tortorici MA, Rey FA and Tautz N (2017) A positive‐strand RNA virus uses alternative protein‐protein interactions within a viral protease/cofactor complex to switch between RNA replication and virion morphogenesis. PLoS Pathog 13, e1006134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Anand K, Palm GJ, Mesters JR, Siddell SG, Ziebuhr J and Hilgenfeld R (2002) Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra α‐helical domain. EMBO J 21, 3213–3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Guarné A, Tormo J, Kirchweger R, Pfistermueller D, Fita I and Skern T (1998) Structure of the foot‐and‐mouth disease virus leader protease: a papain‐like fold adapted for self‐processing and eIF4G recognition. EMBO J 17, 7469–7479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lei J, Mesters JR, Drosten C, Anemüller S, Ma Q and Hilgenfeld R (2014) Crystal structure of the papain‐like protease of MERS coronavirus reveals unusual, potentially druggable active‐site features. Antiviral Res 109, 72–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lei J and Hilgenfeld R (2016) Structural and mutational analysis of the interaction between the Middle‐East respiratory syndrome coronavirus (MERS‐CoV) papain‐like protease and human ubiquitin. Virol Sin 31, 288–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Asselin‐Paturel C and Trinchieri G (2005) Production of type I interferons: plasmacytoid dendritic cells and beyond. J Exp Med 202, 461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lazear HM, Nice TJ and Diamond MS (2015) Interferon‐λ: immune functions at barrier surfaces and beyond. Immunity 43, 15–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ivashkiv LB and Donlin LT (2014) Regulation of type I interferon responses. Nat Rev Immunol 14, 36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Takeuchi O and Akira S (2009) Innate immunity to virus infection. Immunol Rev 227, 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kawai T and Akira S (2009) The roles of TLRs, RLRs and NLRs in pathogen recognition. Int Immunol 21, 317–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jensen S and Thomsen AR (2012) Sensing of RNA viruses: a review of innate immune receptors involved in recognizing RNA virus invasion. J Virol 86, 2900–2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Melchjorsen J, Jensen SB, Malmgaard L, Rasmussen SB, Weber F, Bowie AG, Matikainen S and Paludan SR (2005) Activation of innate defense against a paramyxovirus is mediated by RIG‐I and TLR7 and TLR8 in a cell‐type‐specific manner. J Virol 79, 12944–12951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Egli A, Santer DM, O'Shea D, Tyrrell DL and Houghton M (2014) The impact of the interferon‐lambda family on the innate and adaptive immune response to viral infections. Emerg Microbes Infect 3, e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Davis BK, Wen H and Ting JP (2011) The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol 29, 707–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kawai T and Akira S (2010) The role of pattern‐recognition receptors in innate immunity: update on Toll‐like receptors. Nat Immunol 11, 373–384. [DOI] [PubMed] [Google Scholar]
- 38. Botos I, Segal DM and Davies DR (2011) The structural biology of Toll‐like receptors. Structure 19, 447–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hornung V, Rothenfusser S, Britsch S, Krug A, Jahrsdörfer B, Giese T, Endres S and Hartmann G (2002) Quantitative expression of toll‐like receptor 1‐10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J Immunol 168, 4531–4537. [DOI] [PubMed] [Google Scholar]
- 40. Liu L, Botos I, Wang Y, Leonard JN, Shiloach J, Segal DM and Davies DR (2008) Structural basis of toll‐like receptor 3 signaling with double‐stranded RNA. Science 320, 379–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K et al (2003) Role of adaptor TRIF in the MyD88‐independent toll‐like receptor signaling pathway. Science 301, 640–643. [DOI] [PubMed] [Google Scholar]
- 42. Ono K, Ohtomo T, Sato S, Sugamata Y, Suzuki M, Hisamoto N, Ninomiya‐Tsuji J, Tsuchiya M and Matsumoto K (2001) An evolutionarily conserved motif in the TAB 1 C‐terminal region is necessary for interaction with and activation of TAK1 MAPKKK. J Biol Chem 276, 24396–24400. [DOI] [PubMed] [Google Scholar]
- 43. Cheung PC, Nebreda AR and Cohen P (2004) TAB 3, a new binding partner of the protein kinase TAK1. Biochem J 378, 27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Besse A, Lamothe B, Campos AD, Webster WK, Maddineni U, Lin SC, Wu H and Darnay BG (2007) TAK1‐dependent signaling requires functional interaction with TAB 2/TAB 3. J Biol Chem 282, 3918–3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang C, Deng L, Hong M, Akkaraju GR, Inoue J and Chen ZJ (2001) TAK1 is a ubiquitin‐dependent kinase of MKK and IKK. Nature 412, 346–351. [DOI] [PubMed] [Google Scholar]
- 46. Tseng PH, Matsuzawa A, Zhang W, Mino T, Vignali DA and Karin M (2010) Different modes of ubiquitination of the adaptor TRAF3 selectively activate the expression of type I interferons and proinflammatory cytokines. Nat Immunol 11, 70–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhao T, Yang L, Sun Q, Arguello M, Ballard DW, Hiscott J and Lin R (2007) The NEMO adaptor bridges the nuclear factor‐κB and interferon regulatory factor signaling pathways. Nat Immunol 8, 592–600. [DOI] [PubMed] [Google Scholar]
- 48. Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM and Maniatis T (2003) IKKε and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol 4, 491–496. [DOI] [PubMed] [Google Scholar]
- 49. tenOever BR, Sharma S, Zou W, Sun Q, Grandvaux N, Julkunen I, Hemmi H, Yamamoto M, Akira S, Yeh WC et al (2004) Activation of TBK1 and IKKε kinases by vesicular stomatitis virus infection and the role of viral ribonucleoprotein in the development of interferon antiviral immunity. J Virol 78, 10636–10649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Medzhitov R, Preston‐Hurlburt P, Kopp E, Stadlen A, Chen C, Ghosh S and Janeway CA Jr (1998) MyD88 is an adaptor protein in the hToll/IL‐1 receptor family signaling pathways. Mol Cell 2, 253–258. [DOI] [PubMed] [Google Scholar]
- 51. Kawai T and Akira S (2008) Toll‐like receptor and RIG‐I‐like receptor signaling. Ann N Y Acad Sci 1143, 1–20. [DOI] [PubMed] [Google Scholar]
- 52. Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S and Fujita T (2004) The RNA helicase RIG‐I has an essential function in double‐stranded RNA‐induced innate antiviral responses. Nat Immunol 5, 730–737. [DOI] [PubMed] [Google Scholar]
- 53. Kang DC, Gopalkrishnan RV, Wu Q, Jankowsky E, Pyle AM and Fisher PB (2002) mda‐5: an interferon‐inducible putative RNA helicase with double‐stranded RNA‐dependent ATPase activity and melanoma growth‐suppressive properties. Proc Natl Acad Sci USA 99, 637–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rothenfusser S, Goutagny N, DiPerna G, Gong M, Monks BG, Schoenemeyer A, Yamamoto M, Akira S and Fitzgerald KA (2005) The RNA helicase Lgp2 inhibits TLR‐independent sensing of viral replication by retinoic acid‐inducible gene‐I. J Immunol 175, 5260–5268. [DOI] [PubMed] [Google Scholar]
- 55. Childs K, Randall R and Goodbourn S (2012) Paramyxovirus V proteins interact with the RNA Helicase LGP2 to inhibit RIG‐I‐dependent interferon induction. J Virol 86, 3411–3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Deddouche S, Goubau D, Rehwinkel J, Chakravarty P, Begum S, Maillard PV, Borg A, Matthews N, Feng Q, van Kuppeveld FJ et al (2014) Identification of an LGP2‐associated MDA5 agonist in picornavirus‐infected cells. Elife 3, e01535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bruns AM, Leser GP, Lamb RA and Horvath CM (2014) The innate immune sensor LGP2 activates antiviral signaling by regulating MDA5‐RNA interaction and filament assembly. Mol Cell 55, 771–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ et al (2006) Differential roles of MDA5 and RIG‐I helicases in the recognition of RNA viruses. Nature 441, 101–105. [DOI] [PubMed] [Google Scholar]
- 59. Cao X, Ding Q, Lu J, Tao W, Huang B, Zhao Y, Niu J, Liu YJ and Zhong J (2015) MDA5 plays a critical role in interferon response during hepatitis C virus infection. J Hepatol 62, 771–778. [DOI] [PubMed] [Google Scholar]
- 60. Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O and Akira S (2005) IPS‐1, an adaptor triggering RIG‐I‐ and Mda5‐mediated type I interferon induction. Nat Immunol 6, 981–988. [DOI] [PubMed] [Google Scholar]
- 61. Cai X, Chen J, Xu H, Liu S, Jiang QX, Halfmann R and Chen ZJ (2014) Prion‐like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell 156, 1207–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S et al (2007) TRIM25 RING‐finger E3 ubiquitin ligase is essential for RIG‐I‐mediated antiviral activity. Nature 446, 916–920. [DOI] [PubMed] [Google Scholar]
- 63. Saha SK, Pietras EM, He JQ, Kang JR, Liu SY, Oganesyan G, Shahangian A, Zarnegar B, Shiba TL, Wang Y et al (2006) Regulation of antiviral responses by a direct and specific interaction between TRAF3 and Cardif. EMBO J 25, 3257–3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yoshida R, Takaesu G, Yoshida H, Okamoto F, Yoshioka T, Choi Y, Akira S, Kawai T, Yoshimura A and Kobayashi T (2008) TRAF6 and MEKK1 play a pivotal role in the RIG‐I‐like helicase antiviral pathway. J Biol Chem 283, 36211–36220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ting JP, Lovering RC, Alnemri ES, Bertin J, Boss JM, Davis BK, Flavell RA, Girardin SE, Godzik A, Harton JA et al (2008) The NLR gene family: a standard nomenclature. Immunity 28, 285–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Allen IC, Scull MA, Moore CB, Holl EK, McElvania‐TeKippe E, Taxman DJ, Guthrie EH, Pickles RJ and Ting JP (2009) The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 30, 556–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Rajan JV, Rodriguez D, Miao EA and Aderem A (2011) The NLRP3 inflammasome detects encephalomyocarditis virus and vesicular stomatitis virus infection. J Virol 85, 4167–4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Burdette D, Haskett A, Presser L, McRae S, Iqbal J and Waris G (2012) Hepatitis C virus activates interleukin‐1β via caspase‐1‐inflammasome complex. J Gen Virol 93, 235–246. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 69. Strober W and Watanabe T (2011) NOD2, an intracellular innate immune sensor involved in host defense and Crohn's disease. Mucosal Immunol 4, 484–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sabbah A, Chang TH, Harnack R, Frohlich V, Tominaga K, Dube PH, Xiang Y and Bose S (2009) Activation of innate immune antiviral responses by Nod2. Nat Immunol 10, 1073–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Abbott DW, Yang Y, Hutti JE, Madhavarapu S, Kelliher MA and Cantley LC (2007) Coordinated regulation of Toll‐like receptor and NOD2 signaling by K63‐linked polyubiquitin chains. Mol Cell Biol 27, 6012–6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Schultheiss T, Kusov YY and Gauss‐Müller V (1994) Proteinase 3C of hepatitis A virus (HAV) cleaves the HAV polyprotein P2‐P3 at all sites including VP1/2A and 2A/2B. Virology 198, 275–281. [DOI] [PubMed] [Google Scholar]
- 73. Probst C, Jecht M and Gauss‐Müller V (1999) Intrinsic signals for the assembly of hepatitis A virus particles. Role of structural proteins VP4 and 2A. J Biol Chem 274, 4527–4531. [DOI] [PubMed] [Google Scholar]
- 74. Gitlin L, Barchet W, Gilfillan S, Cella M, Beutler B, Flavell RA, Diamond MS and Colonna M (2006) Essential role of mda‐5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc Natl Acad Sci USA 103, 8459–8464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Oshiumi H, Okamoto M, Fujii K, Kawanishi T, Matsumoto M, Koike S and Seya T (2011) The TLR3/TICAM‐1 pathway is mandatory for innate immune responses to poliovirus infection. J Immunol 187, 5320–5327. [DOI] [PubMed] [Google Scholar]
- 76. Wang B, Xi X, Lei X, Zhang X, Cui S, Wang J, Jin Q and Zhao Z (2013) Enterovirus 71 protease 2Apro targets MAVS to inhibit anti‐viral type I interferon responses. PLoS Pathog 9, e1003231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Feng Q, Langereis MA, Lork M, Nguyen M, Hato SV, Lanke K, Emdad L, Bhoopathi P, Fisher PB, Lloyd RE et al (2014) Enterovirus 2Apro targets MDA5 and MAVS in infected cells. J Virol 88, 3369–3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lind K, Svedin E, Domsgen E, Kapell S, Laitinen O, Moll M and Flodström‐Tullberg M (2016) Coxsackievirus counters the host innate immune response by blocking type III interferon expression. J Gen Virol 97, 1–12. [DOI] [PubMed] [Google Scholar]
- 79. Falah N, Montserret R, Lelogeais V, Schuffenecker I, Lina B, Cortay JC and Violot S (2012) Blocking human enterovirus 71 replication by targeting viral 2A protease. J Antimicrob Chemother 67, 2865–2869. [DOI] [PubMed] [Google Scholar]
- 80. Barral PM, Morrison JM, Drahos J, Gupta P, Sarkar D, Fisher PB and Racaniello VR (2007) MDA‐5 is cleaved in poliovirus‐infected cells. J Virol 81, 3677–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Mukherjee A, Morosky SA, Delorme‐Axford E, Dybdahl‐Sissoko N, Oberste MS, Wang T and Coyne CB (2011) The coxsackievirus B 3C protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling. PLoS Pathog 7, e1001311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lu J, Yi L, Zhao J, Yu J, Chen Y, Lin MC, Kung HF and He ML (2012) Enterovirus 71 disrupts interferon signaling by reducing the level of interferon receptor 1. J Virol 86, 3767–3776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Liu Y, Zhang Z, Zhao X, Yu R, Zhang X, Wu S, Liu J, Chi X, Song X, Fu L et al (2014) Enterovirus 71 inhibits cellular type I interferon signaling by downregulating JAK1 protein expression. Viral Immunol 27, 267–276. [DOI] [PubMed] [Google Scholar]
- 84. Wang C, Sun M, Yuan X, Ji L, Jin Y, Cardona CJ and Xing Z (2017) Enterovirus 71 suppresses interferon responses by blocking Janus kinase (JAK)/signal transducer and activator of transcription (STAT) signaling through inducing karyopherin‐α1 degradation. J Biol Chem 292, 10262–10274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kerekatte V, Keiper BD, Badorff C, Cai A, Knowlton KU and Rhoads RE (1999) Cleavage of Poly(A)‐binding protein by coxsackievirus 2A protease in vitro and in vivo: another mechanism for host protein synthesis shutoff? J Virol 73, 709–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Novoa I and Carrasco L (1999) Cleavage of eukaryotic translation initiation factor 4G by exogenously added hybrid proteins containing poliovirus 2Apro in HeLa cells: effects on gene expression. Mol Cell Biol 19, 2445–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Glaser W and Skern T (2000) Extremely efficient cleavage of eIF4G by picornaviral proteinases L and 2A in vitro . FEBS Lett 480, 151–155. [DOI] [PubMed] [Google Scholar]
- 88. Blom N, Hansen J, Blaas D and Brunak S (1996) Cleavage site analysis in picornaviral polyproteins: discovering cellular targets by neural networks. Protein Sci 5, 2203–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kusov YY and Gauss‐Müller V (1997) In vitro RNA binding of the hepatitis A virus proteinase 3C (HAV 3Cpro) to secondary structure elements within the 5′ terminus of the HAV genome. RNA 3, 291–302. [PMC free article] [PubMed] [Google Scholar]
- 90. Binford SL, Maldonado F, Brothers MA, Weady PT, Zalman LS, Meador JW, Matthews DA and Patick AK (2005) Conservation of amino acids in human rhinovirus 3C protease correlates with broad‐spectrum antiviral activity of rupintrivir, a novel human rhinovirus 3C protease inhibitor. Antimicrob Agents Chemother 49, 619–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kuo CJ, Shie JJ, Fang JM, Yen GR, Hsu JT, Liu HG, Tseng SN, Chang SC, Lee CY, Shih SR et al (2008) Design, synthesis, and evaluation of 3C protease inhibitors as anti‐enterovirus 71 agents. Bioorg Med Chem 16, 7388–7398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Lei X, Liu X, Ma Y, Sun Z, Yang Y, Jin Q, He B and Wang J (2010) The 3C protein of enterovirus 71 inhibits retinoid acid‐inducible gene I‐mediated interferon regulatory factor 3 activation and type I interferon responses. J Virol 84, 8051–8061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Papon L, Oteiza A, Imaizumi T, Kato H, Brocchi E, Lawson TG, Akira S and Mechti N (2009) The viral RNA recognition sensor RIG‐I is degraded during encephalomyocarditis virus (EMCV) infection. Virology 393, 311–318. [DOI] [PubMed] [Google Scholar]
- 94. Rui Y, Su J, Wang H, Chang J, Wang S, Zheng W, Cai Y, Wei W, Gordy JT, Markham R et al (2017) Disruption of MDA5‐mediated innate immune responses by the 3C proteins of coxsackievirus A16, coxsackievirus A6, and enterovirus D68. J Virol 91, e00546–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Yang Y, Liang Y, Qu L, Chen Z, Yi M, Li K and Lemon SM (2007) Disruption of innate immunity due to mitochondrial targeting of a picornaviral protease precursor. Proc Natl Acad Sci USA 104, 7253–7258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Qian S, Fan W, Liu T, Wu M, Zhang H, Cui X, Zhou Y, Hu J, Wei S, Chen H et al (2017) Seneca Valley Virus suppresses host type I interferon production by targeting adaptor proteins MAVS, TRIF, and TANK for cleavage. J Virol 91, e00823–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Lei X, Sun Z, Liu X, Jin Q, He B and Wang J (2011) Cleavage of the adaptor protein TRIF by enterovirus 71 3C inhibits antiviral responses mediated by Toll‐like receptor 3. J Virol 85, 8811–8818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Xiang Z, Li L, Lei X, Zhou H, Zhou Z, He B and Wang J (2014) Enterovirus 68 3C protease cleaves TRIF to attenuate antiviral responses mediated by Toll‐like receptor 3. J Virol 88, 6650–6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Qu L, Feng Z, Yamane D, Liang Y, Lanford RE, Li K and Lemon SM (2011) Disruption of TLR3 signaling due to cleavage of TRIF by the hepatitis A virus protease‐polymerase processing intermediate, 3CD. PLoS Pathog 7, e1002169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Wang D, Fang L, Li K, Zhong H, Fan J, Ouyang C, Zhang H, Duan E, Luo R, Zhang Z et al (2012) Foot‐and‐mouth disease virus 3C protease cleaves NEMO to impair innate immune signaling. J Virol 86, 9311–9322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Wang D, Fang L, Wei D, Zhang H, Luo R, Chen H, Li K and Xiao S (2014) Hepatitis A virus 3C protease cleaves NEMO to impair induction of beta interferon. J Virol 88, 10252–10258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Guo B and Cheng G (2007) Modulation of the interferon antiviral response by the TBK1/IKKi adaptor protein TANK. J Biol Chem 282, 11817–11826. [DOI] [PubMed] [Google Scholar]
- 103. Chariot A, Leonardi A, Muller J, Bonif M, Brown K and Siebenlist U (2002) Association of the adaptor TANK with the IκB kinase (IKK) regulator NEMO connects IKK complexes with IKKε and TBK1 kinases. J Biol Chem 277, 37029–37036. [DOI] [PubMed] [Google Scholar]
- 104. Deng L, Wang C, Spencer E, Yang L, Braun A, You J, Slaughter C, Pickart C and Chen ZJ (2000) Activation of the IκB kinase complex by TRAF6 requires a dimeric ubiquitin‐conjugating enzyme complex and a unique polyubiquitin chain. Cell 103, 351–361. [DOI] [PubMed] [Google Scholar]
- 105. Wang W, Huang X, Xin HB, Fu M, Xue A and Wu ZH (2015) TRAF Family Member‐associated NF‐κB activator (TANK) inhibits genotoxic nuclear factor κB activation by facilitating deubiquitinase USP10‐dependent deubiquitination of TRAF6 ligase. J Biol Chem 290, 13372–13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Huang L, Liu Q, Zhang L, Zhang Q, Hu L, Li C, Wang S, Li J, Zhang Y, Yu H et al (2015) Encephalomyocarditis virus 3C protease relieves TRAF family member‐associated NF‐κB activator (TANK) inhibitory effect on TRAF6‐mediated NF‐κB signaling through cleavage of TANK. J Biol Chem 290, 27618–27632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Huang L, Xiong T, Yu H, Zhang Q, Zhang K, Li C, Hu L, Zhang Y, Zhang L, Liu Q et al (2017) Encephalomyocarditis virus 3C protease attenuates type I interferon production through disrupting the TANK‐TBK1‐IKKε‐IRF3 complex. Biochem J 474, 2051–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Lei X, Han N, Xiao X, Jin Q, He B and Wang J (2014) Enterovirus 71 3C inhibits cytokine expression through cleavage of the TAK1/TAB 1/TAB 2/TAB 3 complex. J Virol 88, 9830–9841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Levy DE, Marié I, Smith E and Prakash A (2002) Enhancement and diversification of IFN induction by IRF‐7‐mediated positive feedback. J Interferon Cytokine Res 22, 87–93. [DOI] [PubMed] [Google Scholar]
- 110. Lei X, Xiao X, Xue Q, Jin Q, He B and Wang J (2013) Cleavage of interferon regulatory factor 7 by enterovirus 71 3C suppresses cellular responses. J Virol 87, 1690–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Xiang Z, Liu L, Lei X, Zhou Z, He B and Wang J (2015) 3C protease of enterovirus D68 inhibits cellular defense mediated by interferon regulatory factor 7. J Virol 90, 1613–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Fink K and Grandvaux N (2013) STAT2 and IRF9: beyond ISGF3. JAKSTAT 2, e27521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Hung HC, Wang HC, Shih SR, Teng IF, Tseng CP and Hsu JT (2011) Synergistic inhibition of enterovirus 71 replication by interferon and rupintrivir. J Infect Dis 203, 1784–1790. [DOI] [PubMed] [Google Scholar]
- 114. Kuyumcu‐Martinez NM, Van Eden ME, Younan P and Lloyd RE (2004) Cleavage of poly(A)‐binding protein by poliovirus 3C protease inhibits host cell translation: a novel mechanism for host translation shutoff. Mol Cell Biol 24, 1779–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Zhang B, Morace G, Gauss‐Müller V and Kusov Y (2007) Poly(A) binding protein, C‐terminally truncated by the hepatitis A virus proteinase 3C, inhibits viral translation. Nucleic Acids Res 35, 5975–5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Clarke BE, Sangar DV, Burroughs JN, Newton SE, Carroll AR and Rowlands DJ (1985) Two initiation sites for foot‐and‐mouth disease virus polyprotein in vivo . J Gen Virol 66, 2615–2626. [DOI] [PubMed] [Google Scholar]
- 117. de Los Santos T, Diaz‐San Segundo F and Grubman MJ (2007) Degradation of nuclear factor kappa B during foot‐and‐mouth disease virus infection. J Virol 81, 12803–12815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. de los Santos T, Segundo FD, Zhu J, Koster M, Dias CC and Grubman MJ (2009) A conserved domain in the leader proteinase of foot‐and‐mouth disease virus is required for proper subcellular localization and function. J Virol 83, 1800–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Wang D, Fang L, Luo R, Ye R, Fang Y, Xie L, Chen H and Xiao S (2010) Foot‐and‐mouth disease virus leader proteinase inhibits dsRNA‐induced type I interferon transcription by decreasing interferon regulatory factor 3/7 in protein levels. Biochem Biophys Res Commun 399, 72–78. [DOI] [PubMed] [Google Scholar]
- 120. Medina GN, Knudsen GM, Greninger AL, Kloc A, Díaz‐San Segundo F, Rieder E, Grubman MJ, DeRisi JL and de Los Santos T (2017) Interaction between FMDV Lpro and transcription factor ADNP is required for optimal viral replication. Virology 505, 12–22. [DOI] [PubMed] [Google Scholar]
- 121. Wang D, Fang L, Li P, Sun L, Fan J, Zhang Q, Luo R, Liu X, Li K, Chen H et al (2011) The leader proteinase of foot‐and‐mouth disease virus negatively regulates the type I interferon pathway by acting as a viral deubiquitinase. J Virol 85, 3758–3766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Mao AP, Li S, Zhong B, Li Y, Yan J, Li Q, Teng C and Shu HB (2010) Virus‐triggered ubiquitination of TRAF3/6 by cIAP1/2 is essential for induction of interferon‐β (IFN‐β) and cellular antiviral response. J Biol Chem 285, 9470–9476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Neuman BW (2016) Bioinformatics and functional analyses of coronavirus nonstructural proteins involved in the formation of replicative organelles. Antiviral Res 135, 97–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Lei J, Kusov Y and Hilgenfeld R (2017) Nsp3 of coronaviruses: structures and functions of a large multi‐domain protein. Antiviral Res, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Mielech AM, Kilianski A, Baez‐Santos YM, Mesecar AD and Baker SC (2014) MERS‐CoV papain‐like protease has deISGylating and deubiquitinating activities. Virology 450–451, 64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Báez‐Santos YM, Barraza SJ, Wilson MW, Agius MP, Mielech AM, Davis NM, Baker SC, Larsen SD and Mesecar AD (2014) X‐ray structural and biological evaluation of a series of potent and highly selective inhibitors of human coronavirus papain‐like proteases. J Med Chem 57, 2393–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Li Y, Chen M, Cao H, Zhu Y, Zheng J and Zhou H (2013) Extraordinary GU‐rich single‐strand RNA identified from SARS coronavirus contributes an excessive innate immune response. Microbes Infect 15, 88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Li SW, Wang CY, Jou YJ, Huang SH, Hsiao LH, Wan L, Lin YJ, Kung SH and Lin CW (2016) SARS coronavirus papain‐Like protease inhibits the TLR7 signaling pathway through removing Lys63‐linked polyubiquitination of TRAF3 and TRAF6. Int J Mol Sci 17, E678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Békés M, van der Heden van Noort GJ, Ekkebus R, Ovaa H, Huang TT and Lima CD (2016) Recognition of Lys48‐linked di‐ubiquitin and deubiquitinating activities of the SARS coronavirus papain‐like protease. Mol Cell 62, 572–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Sun W, Li Y, Chen L, Chen H, You F, Zhou X, Zhou Y, Zhai Z, Chen D and Jiang Z (2009) ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc Natl Acad Sci USA 106, 8653–8658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Ishikawa H and Barber GN (2008) STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Sun L, Xing Y, Chen X, Zheng Y, Yang Y, Nichols DB, Clementz MA, Banach BS, Li K, Baker SC et al (2012) Coronavirus papain‐like proteases negatively regulate antiviral innate immune response through disruption of STING‐mediated signaling. PLoS One 7, e30802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Chen X, Yang X, Zheng Y, Yang Y, Xing Y and Chen Z (2014) SARS coronavirus papain‐like protease inhibits the type I interferon signaling pathway through interaction with the STING‐TRAF3‐TBK1 complex. Protein Cell 5, 369–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Devaraj SG, Wang N, Chen Z, Chen Z, Tseng M, Barretto N, Lin R, Peters CJ, Tseng CT, Baker SC et al (2007) Regulation of IRF‐3‐dependent innate immunity by the papain‐like protease domain of the severe acute respiratory syndrome coronavirus. J Biol Chem 282, 32208–32221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Wang G, Chen G, Zheng D, Cheng G and Tang H (2011) PLP2 of mouse hepatitis virus A59 (MHV‐A59) targets TBK1 to negatively regulate cellular type I interferon signaling pathway. PLoS One 6, e17192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Zheng D, Chen G, Guo B, Cheng G and Tang H (2008) PLP2, a potent deubiquitinase from murine hepatitis virus, strongly inhibits cellular type I interferon production. Cell Res 18, 1105–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Muñoz‐Fontela C, Macip S, Martínez‐Sobrido L, Brown L, Ashour J, García‐Sastre A, Lee SW and Aaronson SA (2008) Transcriptional role of p53 in interferon‐mediated antiviral immunity. J Exp Med 205, 1929–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Yuan L, Chen Z, Song S, Wang S, Tian C, Xing G, Chen X, Xiao ZX, He F and Zhang L (2015) p53 degradation by a coronavirus papain‐like protease suppresses type I interferon signaling. J Biol Chem 290, 3172–3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Ma‐Lauer Y, Carbajo‐Lozoya J, Hein MY, Müller MA, Deng W, Lei J, Meyer B, Kusov Y, von Brunn B, Bairad DR et al (2016) p53 down‐regulates SARS coronavirus replication and is targeted by the SARS‐unique domain and PLpro via E3 ubiquitin ligase RCHY1. Proc Natl Acad Sci USA 113, E5192–E5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Frieman M, Ratia K, Johnston RE, Mesecar AD and Baric RS (2009) Severe acute respiratory syndrome coronavirus papain‐like protease ubiquitin‐like domain and catalytic domain regulate antagonism of IRF3 and NF‐κB signaling. J Virol 83, 6689–6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Clementz MA, Chen Z, Banach BS, Wang Y, Sun L, Ratia K, Baez‐Santos YM, Wang J, Takayama J, Ghosh AK et al (2010) Deubiquitinating and interferon antagonism activities of coronavirus papain‐like proteases. J Virol 84, 4619–4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Shi CS, Shenderov K, Huang NN, Kabat J, Abu‐Asab M, Fitzgerald KA, Sher A and Kehrl JH (2012) Activation of autophagy by inflammatory signals limits IL‐1β production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol 13, 255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Chen X, Wang K, Xing Y, Tu J, Yang X, Zhao Q, Li K and Chen Z (2014) Coronavirus membrane‐associated papain‐like proteases induce autophagy through interacting with Beclin1 to negatively regulate antiviral innate immunity. Protein Cell 5, 912–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Zhu L, George S, Schmidt MF, Al‐Gharabli SI, Rademann J and Hilgenfeld R (2011) Peptide aldehyde inhibitors challenge the substrate specificity of the SARS‐coronavirus main protease. Antiviral Res 92, 204–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Pillaiyar T, Manickam M, Namasivayam V, Hayashi Y and Jung SH (2016) An overview of severe acute respiratory syndrome‐coronavirus (SARS‐CoV) 3CL protease inhibitors: peptidomimetics and small molecule chemotherapy. J Med Chem 59, 6595–6628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Wang D, Fang L, Shi Y, Zhang H, Gao L, Peng G, Chen H, Li K and Xiao S (2015) Porcine epidemic diarrhea virus 3C‐like protease regulates its interferon antagonism by cleaving NEMO. J Virol 90, 2090–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Zhu X, Fang L, Wang D, Yang Y, Chen J, Ye X, Foda MF and Xiao S (2017) Porcine deltacoronavirus nsp5 inhibits interferon‐β production through the cleavage of NEMO. Virology 502, 33–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Zhu X, Wang D, Zhou J, Pan T, Chen J, Yang Y, Lv M, Ye X, Peng G, Fang L et al (2017) Porcine deltacoronavirus nsp5 antagonizes type I interferon signaling by cleaving STAT2. J Virol 91, e00003–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Lawitz E, Sulkowski MS, Ghalib R, Rodriguez‐Torres M, Younossi ZM, Corregidor A, DeJesus E, Pearlman B, Rabinovitz M, Gitlin N et al (2014) Simeprevir plus sofosbuvir, with or without ribavirin, to treat chronic infection with hepatitis C virus genotype 1 in non‐responders to pegylated interferon and ribavirin and treatment‐naïve patients: the COSMOS randomised study. Lancet 384, 1756–1765. [DOI] [PubMed] [Google Scholar]
- 150. Zeuzem S, Jacobson IM, Baykal T, Marinho RT, Poordad F, Bourlière M, Sulkowski MS, Wedemeyer H, Tam E, Desmond P et al (2014) Retreatment of HCV with ABT‐450/r‐ombitasvir and dasabuvir with ribavirin. N Engl J Med 370, 1604–1614. [DOI] [PubMed] [Google Scholar]
- 151. Sumpter R Jr, Loo YM, Foy E, Li K, Yoneyama M, Fujita T, Lemon SM and Gale M Jr (2005) Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG‐I. J Virol 79, 2689–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R and Tschopp J (2005) Cardif is an adaptor protein in the RIG‐I antiviral pathway and is targeted by hepatitis C virus. Nature 437, 1167–1172. [DOI] [PubMed] [Google Scholar]
- 153. Chen Z, Benureau Y, Rijnbrand R, Yi J, Wang T, Warter L, Lanford RE, Weinman SA, Lemon SM, Martin A et al (2007) GB virus B disrupts RIG‐I signaling by NS3/4A‐mediated cleavage of the adaptor protein MAVS. J Virol 81, 964–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Anggakusuma, Brown RJ, Banda DH, Todt D, Vieyres G, Steinmann E and Pietschmann T (2016) Hepacivirus NS3/4A proteases interfere with MAVS signaling in both their cognate animal hosts and humans: implications for zoonotic transmission. J Virol 90, 10670–10681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Oshiumi H, Miyashita M, Matsumoto M and Seya T (2013) A distinct role of Riplet‐mediated K63‐Linked polyubiquitination of the RIG‐I repressor domain in human antiviral innate immune responses. PLoS Pathog 9, e1003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Li K, Li NL, Wei D, Pfeffer SR, Fan M and Pfeffer LM (2012) Activation of chemokine and inflammatory cytokine response in hepatitis C virus‐infected hepatocytes depends on Toll‐like receptor 3 sensing of hepatitis C virus double‐stranded RNA intermediates. Hepatology 55, 666–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Li K, Foy E, Ferreon JC, Nakamura M, Ferreon AC, Ikeda M, Ray SC, Gale M Jr and Lemon SM (2005) Immune evasion by hepatitis C virus NS3/4A protease‐mediated cleavage of the Toll‐like receptor 3 adaptor protein TRIF. Proc Natl Acad Sci USA 102, 2992–2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Dansako H, Ikeda M and Kato N (2007) Limited suppression of the interferon‐β production by hepatitis C virus serine protease in cultured human hepatocytes. FEBS J 274, 4161–4176. [DOI] [PubMed] [Google Scholar]
- 159. Gagné B, Tremblay N, Park AY, Baril M and Lamarre D (2017) Importin β1 targeting by hepatitis C virus NS3/4A protein restricts IRF3 and NF‐κB signaling of IFNB1 antiviral response. Traffic 18, 362–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Yin Z, Patel SJ, Wang WL, Chan WL, Ranga Rao KR, Wang G, Ngew X, Patel V, Beer D, Knox JE et al (2006) Peptide inhibitors of dengue virus NS3 protease. Part 2: SAR study of tetrapeptide aldehyde inhibitors. Bioorg Med Chem Lett 16, 40–43. [DOI] [PubMed] [Google Scholar]
- 161. Nitsche C, Zhang L, Weigel LF, Schilz J, Graf D, Bartenschlager R, Hilgenfeld R and Klein CD (2017) Peptide‐boronic acid inhibitors of flaviviral proteases: medicinal chemistry and structural biology. J Med Chem 60, 511–516. [DOI] [PubMed] [Google Scholar]
- 162. Boldescu V, Behnam MAM, Vasilakis N and Klein CD (2017) Broad‐spectrum agents for flaviviral infections: dengue, Zika and beyond. Nat Rev Drug Discov 16, 565–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Aguirre S, Maestre AM, Pagni S, Patel JR, Savage T, Gutman D, Maringer K, Bernal‐Rubio D, Shabman RS, Simon V et al (2012) DENV inhibits type I IFN production in infected cells by cleaving human STING. PLoS Pathog 8, e1002934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Yu CY, Chang TH, Liang JJ, Chiang RL, Lee YL, Liao CL and Lin YL (2012) Dengue virus targets the adaptor protein MITA to subvert host innate immunity. PLoS Pathog 8, e1002780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Liu H, Zhang L, Sun J, Chen W, Li S, Wang Q, Yu H, Xia Z, Jin X and Wang C (2017) Endoplasmic reticulum protein SCAP inhibits dengue virus NS2B3 protease by suppressing its K27‐linked polyubiquitylation. J Virol 91, e02234–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Onoguchi K, Onomoto K, Takamatsu S, Jogi M, Takemura A, Morimoto S, Julkunen I, Namiki H, Yoneyama M and Fujita T (2010) Virus‐infection or 5′ppp‐RNA activates antiviral signal through redistribution of IPS‐1 mediated by MFN1. PLoS Pathog 6, e1001012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Yu CY, Liang JJ, Li JK, Lee YL, Chang BL, Su CI, Huang WJ, Lai MM and Lin YL (2015) Dengue virus impairs mitochondrial fusion by cleaving mitofusins. PLoS Pathog 11, e1005350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Gottipati K, Ruggli N, Gerber M, Tratschin JD, Benning M, Bellamy H and Choi KH (2013) The structure of classical swine fever virus Npro: a novel cysteine autoprotease and zinc‐binding protein involved in subversion of type I interferon induction. PLoS Pathog 9, e1003704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Bauhofer O, Summerfield A, Sakoda Y, Tratschin JD, Hofmann MA and Ruggli N (2007) Classical swine fever virus Npro interacts with interferon regulatory factor 3 and induces its proteasomal degradation. J Virol 81, 3087–3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Gottipati K, Holthauzen LM, Ruggli N and Choi KH (2016) Pestivirus Npro directly interacts with interferon regulatory factor 3 monomer and dimer. J Virol 90, 7740–7747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Hilton L, Moganeradj K, Zhang G, Chen YH, Randall RE, McCauley JW and Goodbourn S (2006) The Npro product of bovine viral diarrhea virus inhibits DNA binding by interferon regulatory factor 3 and targets it for proteasomal degradation. J Virol 80, 11723–11732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Chen Z, Rijnbrand R, Jangra RK, Devaraj SG, Qu L, Ma Y, Lemon SM and Li K (2007) Ubiquitination and proteasomal degradation of interferon regulatory factor‐3 induced by Npro from a cytopathic bovine viral diarrhea virus. Virology 366, 277–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Szymanski MR, Fiebach AR, Tratschin JD, Gut M, Ramanujam VM, Gottipati K, Patel P, Ye M, Ruggli N and Choi KH (2009) Zinc binding in pestivirus Npro is required for interferon regulatory factor 3 interaction and degradation. J Mol Biol 391, 438–449. [DOI] [PubMed] [Google Scholar]
- 174. Mine J, Tamura T, Mitsuhashi K, Okamatsu M, Parchariyanon S, Pinyochon W, Ruggli N, Tratschin JD, Kida H, Sakoda Y et al (2015) The N‐terminal domain of Npro of classical swine fever virus determines its stability and regulates type I IFN production. J Gen Virol 96, 1746–1756. [DOI] [PubMed] [Google Scholar]
- 175. Fiebach AR, Guzylack‐Piriou L, Python S, Summerfield A and Ruggli N (2011) Classical swine fever virus Npro limits type I interferon induction in plasmacytoid dendritic cells by interacting with interferon regulatory factor 7. J Virol 85, 8002–8011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Doceul V, Charleston B, Crooke H, Reid E, Powell PP and Seago J (2008) The Npro product of classical swine fever virus interacts with IκBα, the NF‐κB inhibitor. J Gen Virol 89, 1881–1889. [DOI] [PubMed] [Google Scholar]
- 177. Tan CW, Lai JK, Sam IC and Chan YF (2014) Recent developments in antiviral agents against enterovirus 71 infection. J Biomed Sci 21, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. de Wit E, van Doremalen N, Falzarano D and Munster VJ (2016) SARS and MERS: recent insights into emerging coronaviruses. Nat Rev Microbiol 14, 523–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Du H, Yin P, Yang X, Zhang L, Jin Q and Zhu G (2015) Enterovirus 71 2C protein inhibits NF‐κB activation by binding to RelA(p65). Sci Rep 5, 14302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Dodd DA, Giddings TH Jr and Kirkegaard K (2001) Poliovirus 3A protein limits interleukin‐6 (IL‐6), IL‐8, and beta interferon secretion during viral infection. J Virol 75, 8158–8165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Noguchi T, Satoh S, Noshi T, Hatada E, Fukuda R, Kawai A, Ikeda S, Hijikata M and Shimotohno K (2001) Effects of mutation in hepatitis C virus nonstructural protein 5A on interferon resistance mediated by inhibition of PKR kinase activity in mammalian cells. Microbiol Immunol 45, 829–840. [DOI] [PubMed] [Google Scholar]
- 182. Taylor DR, Shi ST, Romano PR, Barber GN and Lai MM (1999) Inhibition of the interferon‐inducible protein kinase PKR by HCV E2 protein. Science 285, 107–110. [DOI] [PubMed] [Google Scholar]
- 183. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC and Ferrin TE (2004) UCSF Chimera — a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]