

Summary
While commensal flora is involved in the regulation of immunity, the interplay between cytokine signaling and microbiota in atherosclerosis remains unknown. We found that interleukin (IL)-23 and its downstream target IL-22 restricted atherosclerosis by repressing pro-atherogenic microbiota. Inactivation of IL-23-IL-22 signaling led to deterioration of the intestinal barrier, dysbiosis and expansion of pathogenic bacteria with distinct biosynthetic and metabolic properties, causing systemic increase in pro-atherogenic metabolites such as lipopolysaccharide (LPS) and trimethylamine N-oxide (TMAO). Augmented disease in the absence of the IL-23-IL-22 pathway was mediated in part by pro-atherogenic osteopontin, controlled by microbial metabolites. Microbiota transfer from IL-23 deficient mice accelerated atherosclerosis, whereas microbial depletion or IL-22 supplementation reduced inflammation and ameliorated disease. Our work uncovers the IL-23-IL-22 signaling as a regulator of atherosclerosis that restrains expansion of pro-atherogenic microbiota, and argues for informed use of cytokine blockers to avoid cardiovascular side effects driven by microbiota and inflammation.
Keywords: IL-23, IL-22, atherosclerosis, inflammation, cytokines, microbiome, host-microbe interaction, myeloid cells
eTOC blurb
While anti-cytokine therapies show promising results in autoimmune diseases, its effects on cardiovascular disease development are not well understood. Fatkhullina and colleagues show that inactivation of IL-23-IL-22 signaling leads to deterioration of the intestinal barrier, expansion of pro-atherogenic bacteria and production of metabolites promoting macrophage activation and atherosclerosis.
Graphical Abstract
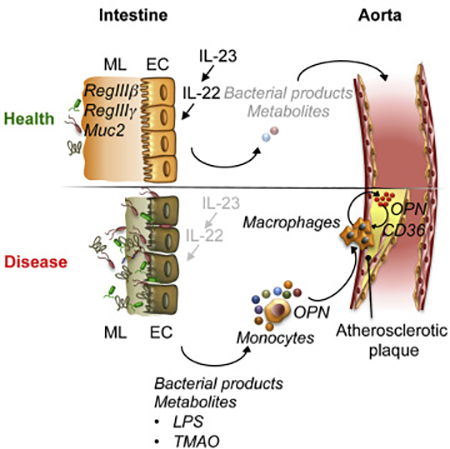
Introduction
Atherosclerosis is a lipid driven, chronic inflammatory disease mediated by activated immune cells in the arterial wall (Galkina and Ley, 2009). A cross talk between the innate and adaptive immune cells, and non-hematopoietic cells in the aortic wall, is mediated by a number of cytokines, which regulate atherogenesis (Ait-Oufella et al., 2011; Hansson and Hermansson, 2011). Elevated concentration of specific cytokines in human and rodent plasma and aortic tissue are linked to atherosclerosis progression (Caligiuri et al., 2003; Han et al., 2010). Anticytokine therapies show promising results in many autoimmune diseases, including inflammatory bowel disease (IBD), psoriasis and arthritis (Gaffen et al., 2014) and, recently, atherosclerosis (Ridker et al., 2017).
Interleukin (IL)-23 is a member of the IL-6-IL-12 family of cytokines and a heterodimer of p19 (unique) and p40 (shared with IL-12) subunits. The IL-23 cytokine axis drives several chronic inflammatory diseases, including arthritis, psoriasis, IBD and cancer; and genetic inactivation or pharmacological blockade of IL-23 generally suppresses disease development (Cua et al., 2003; Hue et al., 2006). IL-23 is a known regulator of IL-17A and IL-22 cytokine production (Gaffen et al., 2014). The role of IL-17A in atherosclerosis has been extensively investigated (Taleb et al., 2010). The mechanisms of IL-22 action in atherosclerosis remain largely unknown, although it has been reported that IL-22 suppresses metabolic disorders and regulates lipid metabolism in the liver and adipose tissue (Behnsen et al., 2014; Wang et al., 2014a). An elevated IL-23 has been reported in aortic tissue of mice and humans with cardiovascular diseases (CVD) (Abbas et al., 2015), while it is reduced in PBMCs of patients with CVD (Khojasteh-Fard et al., 2012). IL-23 has also been implicated in the regulation of cardiac inflammation, as Il23−/− mice display increased injury and adverse remodeling after myocardial infarction (Savvatis et al., 2014). While data in other inflammatory models suggest that IL-23 may potentially function to promote atherosclerosis, the exact role of this cytokine remains unknown.
Microbial and viral components of commensal and pathogenic flora (collectively “microbiome”) are key emerging factors that regulate cytokine production and differentiation of cytokine-producing cells (Ivanov et al., 2009; Khosravi and Mazmanian, 2013). Emerging evidence demonstrates that commensal microbiota have direct effects on immune system activation and inflammatory responses, playing a key role in regulating chronic inflammatory diseases, including rheumatoid arthritis, IBD and, recently, atherosclerosis (Jonsson and Backhed, 2017). Microbiome alterations might systemically impact the progression of distant disease via produced metabolites that increase cardiovascular risk (Jonsson and Backhed, 2017). Therefore, changes in gut microbiota caused by dietary factors, stress, or use of antibiotics, may profoundly impact the development of chronic inflammatory diseases, potentially including atherosclerosis. To date, the mechanisms controlling the interplay between diet, cytokine and immune signaling, and microbiota in atherosclerosis are largely unknown.
Here, we uncovered an atheroprotective role of IL-23-IL-22 signaling. We found that IL23 and IL-22 shape gut microbiota by regulating the production of antimicrobial peptides and restraining the expansion of the semi-invasive bacterial species with pro-atherogenic properties. Metagenomic sequencing and metabolomics along with functional experiments demonstrated that these bacteria promoted atherosclerosis progression particularly by producing lipopolysaccharide (LPS) and components of the trimethylamine N-oxide (TMAO) biosynthesis pathway, upregulating the expression of pro-atherogenic osteopontin (OPN) and facilitating the activation of Ly6Chi monocytes and aortic macrophages. Using genetic, pharmacological, and other approaches, we delineated the mechanisms of cytokine - microbiota-mediated pathway that distantly controls atherosclerosis progression. Our data argues for informed application and further clinical studies of IL-23 and especially IL-22 blockers in individuals with sub-optimal dietary habits and those predisposed to atherosclerosis.
Results
IL-23 deficiency aggravates atherosclerosis
IL-23 cytokine and its downstream target IL-17A have been implicated in the pathogenesis of several autoimmune diseases (Gaffen et al., 2014) and cancer (Grivennikov et al., 2012). The fact that IL-23 regulates pro-atherogenic IL-17A led us to hypothesize that IL-23 might be pathogenic in atherosclerosis. As both IL-23 and its cognate receptor IL-23R are primarily expressed within the hematopoietic compartment, we used a bone marrow (BM) chimera approach to ablate IL-23 signaling in hematopoietic cells in an atherosclerosis-prone mouse model. Age, gender, litter and cage matched atherosclerosis-prone Ldlr−/− mice were lethally irradiated and transplanted with BM from either C57BL/6 (wild type, WT), Il23p19−/− (Il23−/−), or Il23r−/− mice, followed by 16 weeks of a Western Diet (WD) feeding. Atherosclerotic plaque sizes were analyzed in isolated aortic roots stained with Oil Red O. Macroscopic and histological analysis revealed an unexpected significant increase in atherosclerotic plaque sizes in Il23−/− ➔Ldlr−/− mice compared to WT ➔Ldlr−/− controls (Figure 1A-C). No differences in lipid profile, body weight or blood leukocyte counts were detected between cohorts (Figure S1A-C). To corroborate these findings, we tested if IL-23R deficiency phenocopies IL-23 loss. Indeed, Il23r−/− ➔Ldlr−/− mice developed significantly more atherosclerosis compared to WT ➔Ldlr−/− counterparts (Figure S1D). This strong exacerbation of atherosclerosis upon genetic ablation of either IL-23 cytokine or its receptor and suggests an unexpected protective role of IL-23 signaling.
Figure 1. Aggravated atherosclerosis and increased immune cells infiltration in aortas of IL23 deficient mice.

A. Images of aortic arch from Ldlr−/− mice transplanted with Il23−/− or WT BM and fed with WD for 16 weeks. Representative images of aortic root sections (B) and quantitative comparison of atherosclerotic lesion size (C) of Il23−/− ➔Ldlr−/− (n=13) or WT ➔Ldlr−/− (n=13) mice. D. Immune cell composition of aortas isolated from Il23−/− ➔Ldlr−/− (n=13) and WT ➔Ldlr−/− (n=11) was analyzed by flow cytometry. Percentage (left panel) and absolute cell number (right panel) of CD45+ hematopoietic cells, and among them CD11b+, CD11b+CD11c+ and CD11c+ myeloid cells and CD4+ TCRβ+ cells. Relative gene expression in the aortas (E) and intestines (F) of Il23−/− ➔Ldlr−/− (n=10) and WT➔Ldlr−/− (n=10) mice. Gene expression was normalized to RpL32 and then to gene expression in WT➔Ldlr−/− mice. Data are mean ± SEM from at least 3 independent experiments. *p<0.05, **p<0.001, ***p<0.0001. Student’s t-test. See also Figures S1,2,3.
To determine whether IL-23 mediated suppression of atherosclerosis is a general phenomenon, we analyzed disease progression in genetic crosses of Ldlr−/− × Il23−/− and Apoe−/− × Il23−/− mice. Similar to BM transplanted mice, both Ldlr−/− × Il23−/− and Apoe−/− × Il23−/− mice developed significantly larger atherosclerotic lesions (Figure S1E, F). Therefore, the atheroprotective action of IL-23 is general, not limited to the BM chimera approach and is observed in two separate mouse models of atherosclerosis.
Flow cytometry and immunofluorescence revealed increased accumulation of multiple types of hematopoietic cells (CD45+), including CD4+ T cells (TCRβ+CD4+) and myeloid cells subsets (CD11b+, CD11c+, CD11b+CD11c+) in aortas of Il23−/− ➔Ldlr−/− mice (Figure 1D and S2A-B). Additionally, Q-RT-PCR analysis of inflammatory gene expression showed a significant upregulation of Il1b, Tnf, the chemokines Ccl2, Ccl5, Cxcl1 as well Cd36 genes in the aortas of Il23−/− ➔Ldlr−/− mice compared to WT➔Ldlr−/− controls (Figure 1E). Since IL-23 is known to regulate the production of IL-17A by T helper and ILC3 cells, we assayed IL-17A by Q-RT-PCR and intracellular cytokine staining. We did not find any differences in IL-17A between Il23−/−➔Ldlr−/− and WT➔Ldlr−/− mice (Figure 1E and Figure S2C) suggesting that IL-23 may be dispensable for its expression in the atherosclerotic aorta. As previous studies have demonstrated that IL-23 is required for regulating intestinal inflammation (Ahern et al., 2010), we examined inflammatory gene expression in the intestine of mice with atherosclerosis. Consistently, we found a reduction of Il1b, Tnf Ccl2, Ccl5, Cxcl1 and Il22 gene expression in the intestines of Il23−/−➔Ldlr−/− mice compared to WT ➔Ldlr−/− controls (Figure 1F and Figure S2D). Overall, these findings suggest that IL-23 signaling suppresses atherosclerosis.
Inactivation of IL-22 exacerbates atherosclerosis
As IL-23 is also known to regulate IL-22 expression (Zheng et al., 2008), and IL-22 was downregulated in Il23−/− ➔Ldlr−/− mice, we next asked if IL-22 also protects from atherosclerosis. We found strong exacerbation of atherosclerosis in Il22−/− ➔Ldlr−/− mice compared to WT ➔Ldlr−/− littermate and cagemate WD-fed controls (Figure 2A-C). Il22−/− ➔Ldlr−/− mice were characterized by an increased accumulation of T cells and myeloid cells (Figure 2D and S2E, F) in the aortas, and by an upregulation of inflammatory gene expression in aortas and intestines (Figure 2E-F). Transfer of IL-22 deficient BM resulted in strong downregulation of intestinal IL-22 (Figure S2G). We also observed that IL-22 producing CD90.2+ ILC3 and T cells were almost exclusively donor-derived (Figure S2H), thereby excluding incomplete BM repopulation or IL-22 production by residual radio-resistant cells. In agreement with our observation of enhanced atherosclerosis in Il23−/−➔Ldlr−/− and Il22−/−➔Ldlr−/− mice, we found an increased number of apoptotic cells (Figure S3A) and smooth muscle cells (Figure S3B) in the aortas. No difference was detected in tissue repair macrophage genes, IL-12 or lipid transporters expression (Figure S3C-E).
Figure 2. Ablation of IL-22 exacerbates atherosclerosis and IL-22 administration suppresses the disease development in IL-23 deficient mice.

A. Images of aortic arch from Ldlr−/− mice transplanted with Il22−/− or WT BM and fed with WD for 16 weeks. Representative images of aortic root sections (B) and quantitative comparison of atherosclerotic lesion size of Il22−/− ➔Ldlr−/− (n=16) or WT➔Ldlr−/− (n=12) mice (C). D. Immune cell composition of aortas isolated from Il22−/− ➔Ldlr−/− (n=7) and WT➔Ldlr−/− (n=5) was analyzed by flow cytometry. Percentage (left panel) and absolute cell number (right panel) of CD45+ hematopoietic cells, and among them CD11b+, CD11b+CD11c+ and CD11c+ myeloid cells and CD4+ TCRβ+ cells. Relative gene expression in the aortas (E) and intestines (F) of Il22−/− ➔Ldlr−/− (n=10) or WT➔Ldlr−/− (n=10) mice was normalized to RpL32 and then to gene expression in WT➔Ldlr−/− mice. G. Scheme of experiment. Il23−/− ➔Ldlr−/− and WT➔Ldlr−/− were administered with mIL-22-Ig for 4 weeks starting at 8 weeks of WD feeding. H. Representative images of aortic root sections and quantitative comparison of atherosclerotic lesion size from Il23−/− ➔Ldlr−/− (n=13 (control), n=12 (mIL-22-Ig)) and WT➔Ldlr−/− (n=9 (control), n=8 (mIL-22-Ig)) mice treated with mIL-22-Ig or control. ***p<0.0001; *p<0.05. Data are mean ± SEM from 3 independent experiments. *p<0.05, **p<0.001, ***p<0.0001. Student’s t-test. I. IF representative image from 3 independent experiments of aortic root sections from Il23−/−➔Ldlr−/− or WT➔Ldlr−/− mice after mIL-22-Ig treatment demonstrates the reduction of CD11b+, CD11c+, and CD11b+CD11c+ myeloid cells. A-adventitia, M-media, P-atherosclerotic plaque. See also Figure S2,3.
To examine whether IL-23 may restrain atherosclerosis via direct induction of IL-22, we performed a “gain-of-function” experiment, testing whether in vivo supplementation of IL-22 would rectify the pro-atherogenic effects of IL-23 deficiency. We administered IL-22 (mIL-22-Ig) to Il23−/− ➔Ldlr−/− and WT ➔Ldlr−/− mice (Figure 2G) and observed that Il23−/− ➔Ldlr−/− mice receiving mIL-22-Ig developed smaller atherosclerotic lesions compared to controls, while the effect in mIL-22-Ig treated WT ➔Ldlr−/− mice was less prominent (Figure 2H-I). Collectively, these data implicate IL-22 as an immediate IL-23 target in atherosclerosis.
Inactivation of IL-23 or IL-22 drives dysbiosis and pro-atherogenic microbiota alterations
WD can alter microbiota and induce the outgrowth of potentially pathogenic species. Inflammatory cytokines, including IL-23 and IL-22, are known to regulate the function of intestinal epithelial cells and their antimicrobial responses (Zheng et al., 2008). Therefore, we tested whether IL-23 or IL-22 deficiency altered the microbiome and epithelial homeostasis in WD-fed, atherosclerosis-prone mice. We found that intestines of WD-fed Il23−/− ➔Ldlr−/− and Il22−/−➔Ldlr−/− mice displayed a significant reduction of the anti-microbial peptides Reg3b and Reg3g expression compared to WT ➔Ldlr−/− controls (Figure 3A). Immunostaining revealed RegIIIγ downregulation as well as a strong reduction in Mucin2, a key component of mucus (Figure 3B). Ex vivo en face YoYo1 dye DNA staining of whole mount colon and scanning electron microscopy (SEM) demonstrated elevated numbers of bacteria in close proximity to gut epithelium in Il23-/ ➔Ldlr−/− and Il22−/−➔Ldlr−/−, but not in WT ➔Ldlr−/− mice (Figure S4A). No significant difference was found in gene expression of tight junction proteins or intestinal permeability (data not shown). Therefore, IL-23 or IL-22 ablation in WD-fed mice reduces protective, anti-microbial peptides, and results in altered localization of bacteria.
Figure 3. IL-23 or IL-22 deficiency reduces antimicrobial peptide expression and contributes to microbiome alterations in the gut of atherosclerotic mice.

A. Relative antimicrobial peptides gene expression in the intestines of WT➔Ldlr−/− (n=12), Il23−/− ➔Ldlr−/− (n=12) and Il22−/− ➔Ldlr−/− (n=11) mice was normalized to RpL32 and then to gene expression in WT➔Ldlr−/− mice. Data are mean ± SEM from 3 independent experiments. *p<0.05, **p<0.001, ***p<0.0001. Student’s t-test. B. Representative IHC staining of RegIIIγ in colon tissue of WT➔Ldlr−/−, Il23−/− ➔Ldlr−/− or Il22−/− ➔Ldlr−/− mice (top panel), scale bar is 100μm. Representative mucin staining in colon tissue sections of WT➔Ldlr−/−, Il23−/− ➔Ldlr−/− or Il22−/− ➔Ldlr−/− mice stained for Mucin2 (green) and DAPI (blue) (middle panel), scale bar is 100μm. Mucin layer staining after Carnoy’s Fixation (bottom panel), scale bar is 50μm. Images are representative from 3 independent experiments. C. Microbiome compositions in Il23−/− ➔Ldlr−/− and Il22−/− ➔Ldlr−/− mice compared to WT➔Ldlr−/− cage mate controls as determined by whole metagenome shotgun sequencing followed by principal component analysis (PCA). D. Heat map of cecum microbiome composition in Il23−/− ➔Ldlr−/− or Il22−/− ➔Ldlr−/− and WT➔Ldlr−/− mice as determined by shotgun sequencing. Each column represents a single mouse. Red – upregulated bacterial genes, green – downregulated bacterial genes. E, F. Functional characterization of luminal bacteria from Il23−/− ➔Ldlr−/−, Il22−/− ➔Ldlr−/− and WT➔Ldlr−/− mice as determined by metagenome shotgun sequencing. E. Principal component analysis (PCA) and (F) heat map of enzymatic activities (Enzyme Commission (EC) numbers) of sequenced microbiota G. Relative bacterial 16S rRNA expression in the intestines of Il23−/− ➔Ldlr−/− (n=7), Il22−/− ➔Ldlr−/− (n=7) or WT➔Ldlr−/− (n=6) mice. H. Heat map of tissue adhesive bacteria gene expression in Il23−/− ➔Ldlr−/− and WT➔Ldlr−/− mice as determined by 16S rRNA sequencing. Red – upregulated bacteria, blue – downregulated bacteria. D, F, H. Only statistically significantly different results are shown. *p<0.05. See also Figure S4.
To determine if the exacerbation of atherosclerosis in Il23−/− ➔Ldlr−/− and Il22−/− ➔Ldlr−/− mice is mediated by distinct qualitative and quantitative alterations in gut flora, we performed whole metagenome shotgun sequencing analysis of microbiota littermate and cagemate mice cohoused for the entire length of experiment. While global comparison of cecal luminal microbial communities revealed differences between the microbiomes of Il23−/− ➔Ldlr−/−, Il22−/− ➔Ldlr−/− and WT ➔Ldlr−/− mice at the bacteria species level as demonstrated by principal component analysis (Figure 3C), much greater similarities between Il23−/− ➔Ldlr−/− and Il22−/− ➔Ldlr−/− mice versus controls were found at a higher taxonomical level. In particular, the families Enterobacteriaceae (Klebsiella sp), Prevotellaceae (Prevotella copri) and Lachnospiraceae, Clostridiaceae, Ruminococcaceae (Figure 3D) were enriched in both Il23−/− ➔Ldlr−/− and Il22−/− ➔Ldlr−/− mice. These bacterial families have been suggested to be pro-inflammatory or pro-atherogenic (Elinav et al., 2011; Fu et al., 2015; Gregory et al., 2015; Kelly et al., 2016). We found high similarities in enzyme commission (EC) numbers analysis (enzymatic activities) (Figure 3E, F) of luminal bacteria between Il23−/− ➔Ldlr−/− and Il22−/− ➔Ldlr−/− in contrast to WT➔Ldlr−/− controls. Particularly, it revealed an increased presence of the bacterial gene encoding for choline-trimethylamine lyase, an enzyme, required for trimethylamine (TMA) production (Wang et al., 2015).
To further confirm whether downregulation of anti-microbial peptides and mucus allows bacteria to locate close to intestinal epithelial cells, we analyzed changes in adherent microbiota in isolated washed intestinal tissue. Q-RT-PCR analysis revealed an increase in 16S rRNA in Il23−/− ➔Ldlr−/− and Il22−/− ➔Ldlr−/− mice, indicating increased accumulation of epithelium-adhesive bacteria (Figure 3G). 16S rRNA sequencing of tissue-adhesive microbiota supported the Q-RTPCR observations and demonstrated an overrepresentation of Clostridiaceae, Verrucomicrobiaceae, Lachnospiraceae and other families in the epithelium of Il23−/− ➔Ldlr−/− mice (Figure 3H).
To functionally address whether the progression of atherosclerosis is indeed driven by cytokine-dependent changes in microbiota, we treated randomized Il23−/− ➔Ldlr−/− and Il22−/− ➔Ldlr−/− mice with a mix of broad-spectrum antibiotics in drinking water for the last 4 weeks of WD feeding (Figure 4A). As anticipated, antibiotics markedly depleted intestinal microbes (Figure S4B) and resulted in a significant reduction in atherosclerotic plaque size in the Il23−/− ➔Ldlr−/− and Il22−/− ➔Ldlr−/− mice, compared to untreated mice of the same genotype (Figure 4B,C). Both groups of antibiotic-treated mice showed a significant reduction in the expression of proinflammatory genes in the aorta and intestine (Figure 4D and S4C). Conversely, “microbiota transfer” experiments revealed that microbial communities isolated from WD-fed Il23−/− ➔Ldlr−/− mice promoted atherosclerosis and inflammatory gene expression in aortas of WT ➔Ldlr−/− mice (Figure 4E-H and S4D). Microbiota transfer also affected intestinal gene expression (Figure S4E) similarly to found in BM transplanted mice (Figure 1F). Importantly, reciprocal transfer of “wild type microbiome” was unable to suppress the disease in Il23−/− ➔Ldlr−/− mice (Figure 4E-G and S4D, F). These data indicate that microbiota from Il23−/− ➔Ldlr−/− mice possesses specific disease-inducing abilities that can be transferred to the wild type host. 16S rRNA sequencing revealed that fecal transplant from Il23−/− ➔Ldlr−/− donors resulted an increased amount of Clostridiaceae, Lachnospiraceae and Ruminococcaceae in WT ➔Ldlr−/− recipients (Figure S5A-C) and reduced anti-microbial peptides expression (Figure S4E), similarly to Il23−/− ➔Ldlr−/− mice. No significant difference was found in Il23−/− ➔Ldlr−/− recipients of WT microbiota (Figure S4F).
Figure 4. Cytokine-mediated alterations in microbiota drive atherosclerosis progression in Il23−/− ➔Ldlr−/− and Il22−/− ➔Ldlr−/− mice.

A. Scheme of experiment. WT➔Ldlr−/−, Il23−/− ➔Ldlr−/− or Il22−/− ➔Ldlr−/− mice fed with WD for 16 weeks were maintained for the last 4 weeks of WD feeding on regular (Reg) or broad-spectrum antibiotic (Abx) containing water. B. Representative images of aortic root sections (left) and (C) quantitative comparison of atherosclerotic lesion size in WT➔Ldlr−/− (n=8 (Reg), n=5 (Abx)), Il23−/− ➔Ldlr−/− (n=9 (Reg), n=6 (Abx)) or Il22−/− ➔Ldlr−/− (n=12 (Reg), n=6 (Abx)) mice maintained on regular or antibiotic water. Data are average from 3 independent experiments. D. Relative gene expression in aortas from Il23−/− ➔Ldlr−/− (n=5) or Il22−/− ➔Ldlr−/− (n=5) mice maintained on antibiotic-containing or regular water was normalized to RpL32 and then to average gene expression in untreated mice of the same genotype. E. Scheme of microbiota transfer experiment. F. Representative images of aortic root sections and quantitative comparison (G) of atherosclerotic lesion size in WT➔Ldlr−/− (n=7–8) or Il23−/− ➔Ldlr−/− (n=7–9) mice that received microbiome from either WT➔Ldlr−/− or Il23−/− ➔Ldlr−/− mice with advanced atherosclerosis. *p<0.05, **p<0.001. Data are average from 3 independent experiments. H. Relative gene expression in the aortas from WT ➔Ldlr−/− mice that received microbiota from WT ➔Ldlr−/− (n=7) or Il23−/− ➔Ldlr−/− (n=8) atherosclerotic mice was normalized to RpL32 and then to average gene expression in WT ➔Ldlr−/− mice reconstituted with WT microbiota. Data are mean ± SEM from 3 independent experiments Student’s t-test. *p<0.05, **p<0.001. See also Figure S 4,5.
Therefore, IL-23 and IL-22 are required to regulate intestinal homeostasis to prevent outgrowth of pathogenic bacteria and excessive accumulation of bacteria in a close proximity to the epithelium. The exacerbated atherosclerosis observed in the absence of IL-23 or IL-22 is driven directly by microbes, as the atherosclerotic phenotype is accompanied by changes in luminal and mucosa-associated microbes, is “transferrable” to WT recipient by microbiota from IL-23-deficient donor, and can be reversed by microbiota ablation.
Inactivation of IL-23 or IL-22 leads to an increase in metabolites with pro-atherogenic properties
Although upregulated bacterial operational taxonomic units (OTUs) were not always identical between Il23−/− ➔Ldlr−/− and Il22−/− ➔Ldlr−/− mice, initial Picrust analysis of 16S rRNA sequencing data suggested that multiple bacterial pathways, were strongly upregulated in both Il23−/− ➔Ldlr−/− and Il22−/− ➔Ldlr−/− mice compared to WT ➔Ldlr−/− controls (data not shown). These bacterial pathways, including LPS biosynthesis, N-glycosylation, and specific lipid biosynthesis, have been suggested to be pro-inflammatory (Hornemann and Worgall, 2013). A protein domains structure prediction, performed on metagenomic sequencing data, demonstrated the upregulation of the LPS biosynthesis as the most significantly changed pathway (Figure 5A-C), therefore, suggesting that the disruption of IL-23-IL-22 signaling leads to the enrichment of bacterial species capable of producing pro-inflammatory metabolites. This enrichment in bacteria and their closer adherence to the epithelium may allow release of the bacterial metabolites into the systemic circulation, thereby promoting atherosclerosis. In agreement with this idea, we found elevated LPS in sera of both IL-23- and IL-22-deficient Ldlr−/− mice (Figure 5D), implying that the expansion of pathogenic flora may contribute to low-grade systemic inflammation. Importantly, administration of mIL-22-Ig, which “rescues” the IL-23 deficient phenotype (Figure 2H), also reduced serum LPS (Figure 5E) along with “normalizing” microbiota (Figure S5D). Consistently, microbiota depletion by antibiotics reduced serum LPS concentration (Figure 5F), while fecal transplant from IL-23 deficient atherosclerotic donors increased it in WT ➔Ldlr−/− mice (Figure 5G).
Figure 5. Bacterial signaling pathways and metabolites in Il23−/− ➔Ldlr−/− and Il22−/− ➔Ldlr−/− mice.

A-C. Protein domain structure prediction for luminal bacteria from Il23−/− ➔Ldlr−/−, Il22−/− ➔Ldlr−/− and WT➔Ldlr−/− mice as determined by metagenome shotgun sequencing. A. Principal component analysis (PCA). B. Heat map. Red – upregulated, green – downregulated domains. Only statistically significantly changed genes are shown. *p<0.05. C. Presence of Lipopolysaccharide assembly protein A gene domain in microbiota of Il23−/− ➔Ldlr−/−, Il22−/− ➔Ldlr−/− and WT➔Ldlr−/− mice. B, C Only statistically significantly different results are shown. D. Serum Endotoxin (LPS) in WT➔Ldlr−/−, Il23−/− ➔Ldlr−/− or Il22−/− ➔Ldlr−/− (n=22–25). E. Serum Endotoxin (LPS) in WT➔Ldlr−/− and Il23−/−➔Ldlr−/− administered with mIL-22-Ig (n=6 and 10 respectively) or control (n=20). F. Serum Endotoxin (LPS) in WT➔Ldlr−/−, Il23−/− ➔Ldlr−/− or Il22−/− ➔Ldlr−/− mice on regular (n=20) and antibiotics (n=7, 10 and 9 respectively) containing water. G. Serum Endotoxin (LPS) in mice after fecal transplant from WT➔Ldlr−/− or Il23−/− ➔Ldlr−/− into WT➔Ldlr−/− (n=6 and 9 respectively) or Il23−/− ➔Ldlr−/− (n=12 and 9 respectively) mice. H. Presence of TMA lyase bacterial gene in microbiota of Il23−/− ➔Ldlr−/−, Il22−/− ➔Ldlr−/− and WT➔Ldlr−/− mice as determined by shotgun sequencing. I. Cumulative scheme demonstrating upregulation and statistical analysis for multiple metabolites of TMAO pathway in serum of WT➔Ldlr−/−, Il23−/− ➔Ldlr−/− or Il22−/− ➔Ldlr−/− mice (circle) and mice after fecal transplant (diamond). J. Serum TMAO Ldlr−/− x Il23+/+ (n=5) and Ldlr−/− x Il23−/− (n=8) mice fed WD with 1% choline. Data are mean ± SEM from 2–3 independent experiments. Student’s t-test. ANOVA for multiple comparisons. *p<0.05, **p<0.001, ***p<0.0001. See also Figure S6.
Next, to characterize additional mechanistic cues originating from the microbiota and promoting atherosclerosis, we performed discovery metabolomics on serum samples from Il23−/−➔Ldlr−/−, Il22−/− ➔Ldlr−/− and WT ➔Ldlr−/− mice fed with WD for 16 weeks. We detected an increased presence of metabolites related to trimethylamine N-oxide (TMAO) production, including phosphatidylcholine, choline, betaine and trimethylamine (TMA) (Figure 5I), as well as others (Figure S6). These results were in line with an increased presence of bacterial genes encoding for choline-trimethylamine lyase (a TMA biosynthesis enzyme) in Il23−/− ➔Ldlr−/− and Il22−/− ➔Ldlr−/− mice (Figure 5H). A similar upregulation of TMAO pathway metabolites was detected in WT Ldlr−/− recipients of fecal microbiota from IL-23-deficient donors (Figure 5I and S6D). The chemical detection of these metabolites was consistent with metagenome shotgun analysis and 16S rRNA sequencing of luminal and adhesive bacteria, which revealed overrepresentation of bacteria associated with TMAO pathway metabolism and increased presence of choline trimethylamine lyase bacterial gene (Figure 3D, H, 5H). Importantly, TMAO serum concentrations were further elevated in Ldlr−/− x Il23−/− mice fed with WD diet containing 1% choline, a direct metabolic precursor, which is converted into TMAO by bacteria dependent mechanisms (Figure 5J).
Overall these data suggest a mechanistic connection between disrupted IL-22-IL-23 cytokine signaling, a reduction in colonic epithelial barrier function, expansion of pathogenic microbiota, elevated bacteria-derived metabolites in the serum, and atherosclerosis progression, perhaps via induction of low-grade inflammation.
Osteopontin upregulation links microbial changes to progression of atherosclerosis
To further determine how the cytokine ablation and dysbiosis affect inflammatory responses in the aortas, we analyzed inflammatory gene expression and found osteopontin (OPN, Spp1) as highly upregulated gene in aortas of Il23−/− ➔Ldlr−/− and Il22−/− ➔Ldlr−/− mice (Figure 6A). OPN, a secreted inflammatory protein, has been implicated in various inflammatory conditions and suggested to play a role in atherosclerosis (Isoda et al., 2003; Kahles et al., 2014; Matsui et al., 2003). We found that OPN protein was highly expressed in CD11b+CD11c- macrophages in the aortas (Figure 6B), while its expression was modest in macrophages in other tissues, including peritoneal cavity or spleen (data not shown).
Figure 6. Osteopontin is required for enhanced atherosclerosis in Il23−/− ➔Ldlr−/− and Il22−/−</P/> ➔Ldlr−/− mice.

A. Relative gene expression of Spp1 in the aortas of WT➔Ldlr−/− (n=12), Il23−/− ➔Ldlr−/− (n=13) or Il22−/− ➔Ldlr−/− (n=13) was normalized to RpL32 and then to gene expression in WT➔Ldlr−/− mice. B. Intracellular OPN staining in myeloid cells isolated from aortas of Il23−/− ➔Ldlr−/− mice. C. Q-PCR gene expression analysis of Spp1 in aortas of Il23−/− ➔Ldlr−/− (n=6) or Il22−/− ➔Ldlr−/− (n=5) mice, treated with regular (Reg) or antibiotic (Abx) water for the last 4 weeks of WD feeding. Normalized to RpL32 and then to average gene expression in untreated mice (n=6 per group) of the same genotype. D. OPN gene expression in WT ➔Ldlr−/− recipients of fecal transplant from WT ➔Ldlr−/− or Il23−/− ➔Ldlr−/− atherosclerotic mice. E. Scheme- WT➔Ldlr−/−, Il23−/− ➔Ldlr−/− or Il22−/− ➔Ldlr−/− mice were administered with anti-OPN-antibody or control every 3 days for the last 2.5 weeks of WD feeding. Representative images of aortic root sections (F) and quantification of atherosclerotic lesion size (G) from WT➔Ldlr−/− (n=7–10), Il23−/− ➔Ldlr−/− (n=8–13) or Il22−/− ➔Ldlr−/− (n=7–11) mice treated with antiOPN-antibody or control (Black – PBS, Blue – IgG1k isotype control). H. Confocal microscopy of myeloid cells in aortic roots of indicated anti-OPN-treated or control mice. I. Relative gene expression in aortas from Il23−/− ➔Ldlr−/− (n=6) and Il22−/− ➔Ldlr−/− (n=4) mice, injected with anti-OPN-antibody or control. Gene expression was normalized to RpL32 gene expression and then normalized to average of gene expression of untreated mice of the same genotype. Data are mean ± SEM and images are representative from 3 independent experiments. Student’s t-test. *p<0.05, **p<0.001, ***p<0.0001.See also Figure S7.
Next, we found that microbiota depletion by antibiotics led to a significant downregulation of Spp1 in Il23−/− ➔Ldlr−/− and Il22−/− ➔Ldlr−/− mice (Figure 6C), while microbiota transfer from Il23−/−➔Ldlr−/− donors induced Spp1 expression in WT ➔Ldlr−/− recipients (Figure 6D), suggesting that a particular constellation of microbial compounds, available in IL-23 deficient mice, is capable to induce OPN. To mechanistically test if OPN is an important mediator of atherosclerosis aggravation, we treated randomized Il23−/− ➔Ldlr−/−, Il22−/− ➔Ldlr−/− and WT ➔Ldlr−/− mice with anti-OPN antibody for the last 2.5 weeks of WD feeding (Figure 6E). OPN neutralization significantly reduced atherosclerosis in both Il23−/− ➔Ldlr−/− and Il22−/− ➔Ldlr−/− mice, bringing disease manifestations to those in WT ➔Ldlr−/− controls (Figure 6F, G). We found a significant reduction in myeloid and T cells in the aortic roots of both Il23−/− ➔Ldlr−/− and Il22−/− ➔Ldlr−/− mice treated with anti-OPN antibody (Figure 6H and S7A-C). This reduction of inflammatory cells was accompanied by downregulation of Il1b, Ccl2 and Ccl5 in anti-OPN-treated Il23−/− ➔Ldlr−/− and Il22−/− ➔Ldlr−/− mice (Figure 6I).
Finally we tested whether metabolites elevated in IL-23 and IL-22 deficient mice could directly promote OPN expression and atherosclerosis progression. We treated mice for 3 weeks with LPS and TMAO and found that combined administration of these metabolites accelerated atherosclerosis development in WD-fed Ldlr−/− mice (Figure 7A, B) along with upregulation of Spp1, Cd36 and Il1b gene expression in aortas (Figure 7C). Similar results were obtained when explanted aortas were stimulated with LPS and TMAO ex vivo (Figure S7D). We further found that acute in vivo administration of LPS and TMAO enhances Spp1 expression in circulating Ly6Chi monocytes (Figure 7D), suggesting that OPN may become upregulated in circulating cells. It is conceivable that these pre-activated monocytes are then recruited to atherosclerotic plaques, where they differentiate into macrophages and produce more OPN in an environment rich in OPN-inducing stimuli.
Figure 7. In vivo administration of LPS and TMAO accelerates atherosclerosis development.
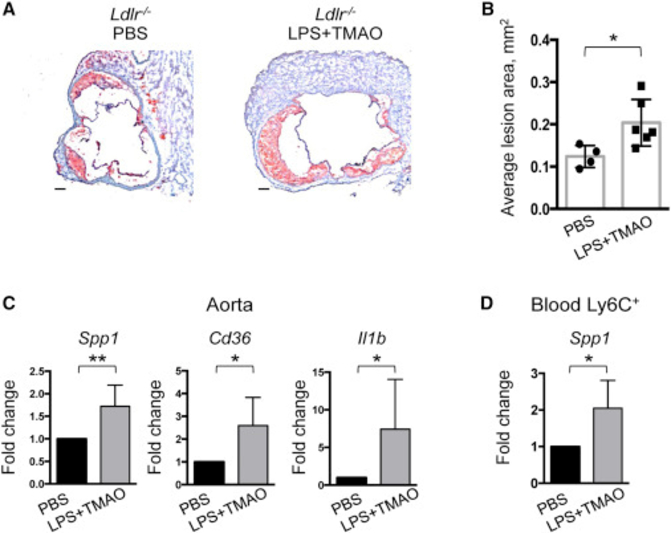
Representative images of aortic root sections (A) and quantitative comparison of atherosclerotic lesion size (B) from Ldlr−/− mice fed with WD for 6 weeks followed by administration with LPS and TMAO or PBS control for 3 weeks every 3 days. C. Spp1, Cd36 and Il1b genes expression in the aortas of Ldlr−/− mice treated with LPS+TMAO or PBS control. D. Spp1 expression in circulating Ly6Chi monocytes in response to LPS and TMAO administration (n=4–6). Gene expression was normalized to RpL32 and then to average gene expression in controls. Images are representative from 2 independent experiments. Data are mean ± SEM. Student’s t-test. *p<0.05, **p<0.001. See also Figure S7.
Together, these data suggest that OPN is an important driver of atherosclerosis and its expression is regulated by pathogenic microbiota and metabolites, which are kept at bay by a functional IL-23-IL-22 pathway. Altogether, a functional atheroprotective IL-23-IL-22 axis maintains intestinal homeostasis and normal host-microbe interactions, and prevents outgrowth of epithelia-associated, pro-atherogenic bacteria. These bacteria deliver pro-atherogenic metabolites, such as LPS and TMAO systemically due to the defective intestinal barrier. These metabolites conspire with additional pro-atherogenic factors to enhance the activation of circulating monocytes and pathogenic inflammatory macrophages promoting atherosclerosis, particularly in OPN-dependent manner.
Discussion
A field connecting the diet-modulated microbiome and metabolic diseases has recently emerged. Microbial alterations have been shown to regulate cytokine production, which is required for host defense against intestinal infections (Belkaid and Hand, 2014). Conversely, cytokine signaling has been also implicated in regulating host-microbiome interactions during infections (Kamada et al., 2013). As atherosclerosis is an inflammatory disease, it could be expected that cytokine blockade, which has been effective in multiple forms of autoimmunity, might be beneficial against this chronic CVD condition as exemplified by the recently completed CANTOS trial (Ridker et al., 2017). However, the fact that cytokines are important regulators of host-microbial interactions provides an alternative possibility, namely that cytokine blockade will alter distinct qualitative and quantitative characteristic of the microbiota, further affecting metabolic parameters. Both of these possibilities were largely unexplored, especially for a cytokine like IL-23, whose role in atherosclerosis remains elusive.
Our work reveals that cytokine signaling, which plays a key role in driving systemic inflammation, is also involved in homeostatic regulation of intestinal epithelial cells, controlling expansion of atherogenic microbiota. We have shown that genetic inactivation of either IL-23 or its receptor, IL-23R, aggravated atherosclerosis progression in a long-term model, which allows microbiota changes to develop and microbiota-driven changes to manifest. In previous studies, systemic expression of IL-23 in a transgenic model induced an inflammatory phenotype and promoted various inflammatory diseases, predisposing to atherosclerosis (Wiekowski et al., 2001). Short-term IL-23 injections have been shown to increase apoptosis in plaques, likely acting in a GM-CSF dependent manner, and it was shown that early lesions could develop unperturbed in the absence of IL-23R (Engelbertsen et al., 2018; Subramanian et al., 2015). In our model genetic inactivation of IL-23 resulted in an increased number of apoptotic cells in atherosclerotic plaques, confirming that apoptosis in the plaque is tightly linked to atherosclerosis progression.
While we originally hypothesized that IL-23 would regulate IL-17A expression in the aorta and thereby would promote atherosclerosis in IL-17A dependent manner, we did not find any differences in IL-17A expression in the aortas of IL-23 or IL-23R deficient mice. Other factors, for instance, IL-1β strongly upregulated in the aortas of Il23−/− ➔Ldlr−/− mice, could in turn promote IL-17A production in the absence of IL-23. On the other hand, we found a significant reduction of IL-22 expression, another cytokine regulated by IL-23, in the intestines of atherosclerotic Il23−/− ➔Ldlr−/− mice. Our data revealed that IL-22 had a strong protective role in atherosclerosis, as Il22−/− ➔Ldlr−/− mice developed exacerbated disease compared to WT ➔Ldlr−/− controls, although a pro-atherogenic role of this cytokine was also previously suggested (Rattik et al., 2015). Administration of IL-22 suppressed the disease in IL-23 deficient mice, implying IL22 as a critical molecule downstream of IL-23. The observed differences in phenotypes may be attributed to several factors, including possible genetic variations between the mice used or differences in the microbiome of non-cohoused and non-littermate animals, supporting the hypothesis that IL-22 has an important microbiome-regulatory role in atherosclerosis.
Cytokine signaling in intestinal epithelial cells has been suggested to modulate microbiota composition (Belkaid and Hand, 2014; Kamada et al., 2013). IL-22 has been found to control epithelial cell proliferation and antimicrobial peptide production, limiting the ability of commensal bacteria to cause inflammation (Behnsen et al., 2014; Sugimoto et al., 2008). Changes in the production of anti-microbial peptides therefore can result in the expansion of bacterial species with potentially pathogenic properties and their localization in direct proximity to intestinal epithelium. In agreement, we found a significant reduction of RegIIIγ and Mucin 2 in Il22−/− ➔Ldlr−/− and Il23−/− ➔Ldlr−/− atherosclerotic mice, which correlated with a high amount of tightly adhesive bacteria including Clostridiaceae and Ruminococcaceae known to contribute to mucus layer degradation (Desai et al., 2016; Tailford et al., 2015).
Quantitative alterations of the microbiome have been associated with diseases, including IBD, metabolic disorders, cancer and atherosclerosis (Elinav et al., 2011; Goodman and Gordon, 2010; Wang et al., 2011). We found that the augmented atherosclerosis progression in WD-fed Il23−/− ➔Ldlr−/− and Il22−/− ➔Ldlr−/− mice was associated with the enrichment in bacteria with previously suggested pro-inflammatory, pro-atherogenic roles (Kelly et al., 2016; Wang et al., 2015). Conversely, potentially protective Odoribacteriaceae were detected in WT ➔Ldlr−/−, but not Il23−/− ➔Ldlr−/− or Il22−/− ➔Ldlr−/− mice. As microbiome alterations could systemically impact distant disease progression via release of metabolites (Postler and Ghosh, 2017; Schroeder and Backhed, 2016), we found that the IL-23-IL-22-microbiota axis was essential for regulating several relevant bacterial functional and metabolic pathways, and thus, atherosclerosis. One large group of these pathways increased the adhesiveness of bacteria, the synthesis of toxic bacterial products such as N-glycans, and the sphingolipid biosynthesis pathway. LPS biosynthesis, however, was the highest among the upregulated pathways. Deterioration of epithelial barrier function and adhesiveness of bacteria to the epithelium, combined with elevated biosynthesis of these metabolites, is likely to functionally contribute to the increased systemic concentration of LPS detected in atherosclerotic mice with IL-23 or IL-22 ablation. Alterations of other pathways, found by metagenomics shotgun sequencing were also confirmed by discovery metabolomics detecting elevated presence of phosphatidylcholine, choline, TMA and trimethylamine-N-oxide (TMAO) in serum of Il23−/− ➔Ldlr−/− mice. Among them, TMAO has been implicated in the pathogenesis of atherosclerosis in humans and mice consuming a choline-rich diet (Wang et al., 2011). Increased TMAO production in the absence of IL-23 pathway was further elevated by feeding with choline rich WD, implying that IL-23-IL-22microbiota-TMAO pathway may be especially important in individuals with different sub-optimal dietary habits. Serum TMAO positively correlated with enrichment for certain bacteria, including the Prevotellaceae, Enterobacteriaceae, Ruminococcaceae and Clostridiaceae, linked to TMAO production and atherosclerosis progression in correlation studies in humans (Brusca et al., 2014; Fu et al., 2015; Gregory et al., 2015; Kelly et al., 2016; Yan et al., 2017). Moreover, metagenomics data revealed increased presence of the bacterial gene encoding for cholinetrimethylamine lyase in IL-23 and IL-22 deficient mice, providing linear connection between dysbiosis, pathogenic metabolites and atherosclerosis. The data obtained from the bacterial pathways analysis may be highly relevant to humans, who are much more diverse in microbes and may harbor pathogenic and protective microbes of different species, but with similar metabolic and biosynthetic profiles.
While metabolites like TMAO and LPS can potentially drive atherosclerosis directly, they can also regulate inflammatory intermediates essential for disease pathogenesis. Indeed, enhanced atherosclerosis in the absence of IL-23-L-22 pathway was accompanied by increased systemic inflammation. We found strong upregulation of osteopontin (Spp1) in circulating Ly6Chi monocytes and CD11b+CD11c- aortic macrophages in the absence of IL-23 or IL-22 signaling. OPN is known to be induced by inflammatory, atherosclerosis-relevant stimuli, such as oxidized lipoproteins (oxLDL) and various inflammatory cytokines (Kahles et al., 2014). OPN regulates the recruitment and activation of both adaptive and innate immune cells in inflammatory diseases and cancer (Isoda et al., 2003; Kahles et al., 2014; Matsui et al., 2003) and particularly could act also on circulating monocytes in an autocrine manner in atherosclerosis. Importance of OPN in control of excessive inflammation and atherosclerosis downstream of microbial alterations was illuminated by reduction in OPN after antibiotic treatment, increase in OPN after microbial transfer from Il23−/− ➔Ldlr−/− donors and reduction in atherosclerosis after OPN neutralization.
Our data, therefore, suggests a model in which inhibition of IL-23 or IL-22 pathways results in alterations in the microbiome, which, coupled with a reduced anti-microbial, protective epithelial layer, leads to elevated serum concentration of metabolites and bacterial products, such as LPS and TMAO, promoting activation of monocytes and aortic macrophages and further progression of atherosclerosis, particularly in OPN-dependent manner. IL-23-IL-22 cytokine signaling therefore controls qualitative and quantitative characteristics of the microbiota and its pro-atherogenic metabolites, and distantly suppresses atherosclerosis via regulation of intestinal homeostasis and inflammation, although we do not exclude an aorta-specific role of these cytokines. Diet and cytokine signaling-induced changes in pro-atherogenic metabolites regulate OPN expression, inflammation and atherosclerosis progression. Our findings will be informative about mechanisms underlying cardiovascular adverse effects, which could be observed upon IL-23 and IL-22 cytokine blockade, especially in subjects with elevated cardiovascular risk factors and/or suboptimal dietary habits (e.g. high carbohydrates, fat or choline containing food). In the long run, inactivation of IL-23-IL-22 axis may exacerbate dormant CVD. As IL-22 is a key cytokine preventing atherosclerosis downstream of IL-23, strategies, which spare IL-22 pathway during cytokine blockade, may be justified. Most importantly, our work demonstrates mechanistic connection between cytokine signaling and host-microbiota interaction in distant regulation of prevalent, diet-driven cardiovascular disease.
STAR Methods
Contact for reagents and resource sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ekaterina Koltsova (Ekaterina.Koltsova@fccc.edu)
Experimental models and subject details
Animals
All animal experiments were approved by the Animal Care Committee at FCCC. Ldlr−/− and Apoe−/− were purchased from Jackson labs. IL-23-, IL-22-deficient mice (Il23p19 referred in the text as Il23−/−, Il22−/−) were provided by Genentech. IL-23R deficient mice were initially generated by G. Trinchieri (NCI/NIH) and described (Grivennikov et al., 2012). Il23−/− were bred with Ldlr−/− and Apoe−/− mice to obtain Ldlr−/− x Il23−/− and Apoe−/− x Il23−/− mice. All mice were on C57BL/6 background and were bred in house. See also Key Resources Table.
Beginning at 6 weeks of age, female and male Ldlr−/− x Il23−/− and Apoe−/− x Il23−/− mice were placed on WD; Ldlr−/− mice were used as bone marrow recipients; Il23−/−, Il23r−/−, Il22−/− were used as donors. After 4 weeks of bone marrow reconstitution, recipients were fed with Western diet (WD) (TD 88137: 15.2% kcal from protein, 42.7% kcal from carbohydrate, 42% kcal from fat, 0.2% cholesterol) https://www.envigo.com/resources/data-sheets/88137.pdf) or custom made WD (TD88137) with 1% choline (Teklad TD160118) for 16 weeks. Mice were housed under specific pathogen-free conditions in an AAALAC-approved barrier facility at Fox Chase Cancer Center (FCCC) and littermate controls were used (each cage contained WT, Il23−/− (or Il23r−/−) and Il22−/− bone marrow transplanted mice). Mice were genotyped by standard PCR protocols.
IL-22 supplementation experiments:
Bone marrow transplanted Il23−/− ➔Ldlr−/− and WT ➔Ldlr−/− mice fed with WD for 8 weeks were injected intraperitoneally (i.p.) every 3 days for 4 weeks with recombinant mouse IL-22 cytokine, kindly provided by Genentech Inc, (50μg per injection) or PBS.
Antibiotic treatment experiments:
At 12 weeks of WD feeding WT➔Ldlr−/−, Il23−/−➔Ldlr−/− and Il22−/−➔Ldlr−/− mice were treated with water containing a mix of broad-spectrum antibiotics: ciprofloxacin 0.2g/L, neomycin 1g/L, vancomycin 0.5g/L, ampicillin 1g/L, metronidazole 0.5g/L, primaxin 0.5g/L.
OPN neutralization experiments:
13.5 weeks WD fed WT➔Ldlr−/−, Il23−/− ➔Ldlr−/− and Il22−/− ➔Ldlr−/− mice were injected intraperitoneally (i.p.) every 3 days for 2.5 weeks with mouse OPN-antibody (purified from hybridoma, clone MPIIIB10, DSHB, University of Iowa) (100μg/mouse per injection) or control represented by either PBS or mouse IgG1k isotype control (clone MOPC-21) in 3 independent experiments.
Miicrobiota transfer experiments:
Microbiota recipients WT➔Ldlr−/− or Il23−/− ➔Ldlr−/− mice were maintained for 8 days with a mix of broad-spectrum antibiotics in drinking water (ciprofloxacin 0.2g/L, neomycin 1g/L, vancomycin 0.5g/L, ampicillin 1g/L, metronidazole 0.5g/L and primaxin 0.5g/L). 24 hours before microbiota transfer, recipient mice were placed on regular water. Microbiota from cecum of WT ➔Ldlr−/− or Il23−/− ➔Ldlr−/− mice fed with WD for 16 weeks was collected, weighted, resuspended at concentration 1g of feces/50ml of PBS and gavaged in PBS in 3 consecutive days after which mice were fed with WD and housed separately for 16 weeks for atherosclerosis studies.
Methods details
Bone marrow transplantation
Recipient Ldlr−/− mice (6–8 weeks old) were irradiated in 2 equal doses of 600 rads 3–4 hours apart. Femurs and tibias of donor mice (WT, Il23r−/−, Il23−/− or Il22−/−) were collected and bone marrow were isolated and resuspended in PBS under sterile conditions. After the second irradiation recipient mice were injected intravenously (i.v.) with 5*106 bone marrow cell suspensions and maintained on water, containing antibiotics (neomycin and polymixin B) during for first 2 weeks post-transplant. Starting 4 weeks after BMT mice were fed with WD for 16 weeks to promote the atherosclerosis development.
Quantification of aortic atherosclerotic lesions
Aortic roots were isolated, frozen in an Optimal Cutting Temperature (O.C.T.) compound (Tissue Tek (Sakura)) on dry ice and stored at −80°C. Five- micrometer frozen tissue sections were cut starting at the aortic valve plane to cover 500μm in intervals of 50μm and stained with Oil Red O/Hematoxylin/Light green as previously described. Images were obtained using Nikon Eclipse 80i microscope with a 4×0.2 NA objective. The lesion area was measured using Fiji software; atherosclerosis lesion area was calculated as average of lesion areas at least 8 sections for each mouse.
Immune cells composition analysis by Flow Cytometry
Aortas were isolated from WT ➔Ldlr−/−, Il23−/− ➔Ldlr−/− and Il22−/− ➔Ldlr−/− mice, cut into small pieces followed by digesting in 2 ml of enzymatic cocktail, containing 450 U/ml collagenase type I, 250 U/ml collagenase type XI, 120 U/ml hyaluronidase type I, 120 U/ml DNAse I in 1x HBSS and incubated in a shaker at 37°C for 55 min. Obtained single cells suspension was stained with following antibodies: CD45-PerCP (30-F11), CD11b-eFluor 450 (M1/70), CD11c-Cy7PE (N418), TCRβ-eFluor 780 (H57–597), CD4-PE (GK1.5) and LIVE/DEAD Yellow fixable dye (Invitrogen) and analyzed by flow cytometry (LSRII, BD Biosciences). For T-cell related cytokine production analysis, single-cell suspensions from aorta were stimulated in complete RPMI 1640 media with PMA+Ionomycin and Brefeldin A for 5h in vitro at 37°C in a CO 2 incubator. For intracellular myeloid cells osteopontin staining, cells were stimulated in complete DMEM media with LPS (100ng/ml) for 4h in the presence of Brefeldin A. After stimulation cells were fixed and permeabilized by Cytofix/Cytoperm kit (BD), and stained with IL-17A-Cy7PE (eBio17B7), IFNγ-APC (XMG1.2) or OPN-APC (2F10) antibody.
Lamina propria lymphocyte (LPL) isolation was performed as described before (Grivennikov et al., 2012). In brief, isolated intestines were removed and placed in ice-cold 5% fetal bovine serum (FBS) in HBSS and incubated twice in 20ml of 5mM EDTA in HBSS for 15 min at 37°C with rotation of 150 rpm. After incubation, IELs were removed by passing through a metal sieve into new tubes and the residual tissues (LPL) were chopped to make it almost homogeneous and digested in 5–6 ml collagenase type VIII (Sigma) (1mg/ml) in 5% FBS, HBSS for 35 min in shaking 37°C incubator. After incubation the soluti on was passed through 70μm cell strainer. Percoll gradient (40%/80%) separation of LPL was performed by centrifugation at 2200rpm for 25 min with slow acceleration and deceleration. LPL immune cells were collected from the interphase of Percoll gradient. Obtained cells suspension was stained with following antibodies: CD45-PerCP (30-F11), CD90.2-eFluor 450 (M1/70), TCRβ-eFluor 700 (H57–597), CD4-APC (GK1.5) and LIVE/DEAD Yellow fixable dye (Invitrogen) and analyzed by flow cytometry (LSRII). For IL-22 cytokine production analysis, single-cell suspensions from the LPL and aorta were stimulated in complete RPMI 1640 media with IL-23 (50ng/mL), IL-2 (5ng/mL) and IL-6 (5ng/mL) recombinant cytokines for 14 hours at 37°C in a CO 2 incubator. Starting at 10 hour stimulation Brefeldin A was added to retain the cytokines within the cell. After stimulation cells were fixed and permeabilized by Cytofix/Cytoperm kit (BD Biosciences), and stained with IL-22-PE (1H8PWSR). Flow cytometry data were analyzed using FlowJo software.
Immunofluorescent and Immunohistochemistry staining analysis
Frozen 5μm aortic root and colon sections were fixing for 10 min in acetone (Sigma-Aldrich) at RT and placed into 1% paraformaldehyde in 100mM sodium phosphate containing 60mM lysine and 7mM sodium periodate (Sigma-Aldrich), pH 7.4 on ice. The sections were incubated with avidin/biotin blocking kit (Vector laboratories) following incubation in a 5% normal goat serum and 1% BSA (Sigma) in PBS. Aortic root sections were stained overnight at 4°C with primary antibodies: hamster anti-CD11c (HL3), rat anti-CD11b-FITC (M1/70), rat anti-CD3 (17A2) and rabbit smooth muscle actin, followed by staining with secondary antibodies for 1 h at RT: goat anti-FITC Alexa Fluor 488 (Molecular Probes), goat anti-hamster IgG DyLight 649 (Jackson Immunoresearch), goat anti-rat IgG Alexa Fluor 568 (Molecular Probes), and donkey anti-rabbit IgG Alexa Fluor 568.
Colon tissues without contents were fixed in Carnoy’s fixative (60% ethanol, 30% chloroform, 10% glacial acetic acid) overnight, paraffin-embedded and stained for Mucin2 with rabbit antiMuc2 (Santa Cruz), followed by goat anti-rabbit Alexa 488 (Life Technologies) secondary antibody staining.
RegIIIγ and Mucin staining were performed on colon tissues fixed in 4% Paraformaldehyde overnight and 30% Sucrose at 4°C for 12h and frozen in O.C.T. compound (Tissue Tek (Sakura)) on dry ice.
Sections were counterstained with DAPI and embedded in Prolong Gold. Images were acquired on a Leica SP8 DM6000 inverted confocal microscope using HCX PLAPO 20x and 40x oil-immersion objectives at 405 nm, 488 nm, 563 nm and 633 nm excitation wavelength. Imaris Software was employed to adjust brightness and one-step smoothing on all images in parallel. Immunohistochemistry staining for RegIIIγ was performed on frozen 5μm colon sections that were fixing for 10 min in acetone (Sigma-Aldrich). Fixed sections were incubated in 3% H2O2 solution in PBS at room temperature for 10 min to block endogenous peroxidase activity following by incubation with 5% normal goat serum in PBS for 1 hour at room temperature. Slides were stained overnight at 4°C with primary a ntibodies: rabbit anti-REGIIIγ (Invitrogen) followed by staining with biotinylated secondary goat antibodies for 1h at RT. Streptavidin-HRP (BD, Pharmingen) was added for 30 min. Sections were developed with DAB substrate (0.05% DAB and 0.015% H2O2 in PBS) for 3–5 min and counterstained with Hematoxylin (1–2min) and then immerged in bluing for 30 seconds. Tissue sections were dehydrated through 3 changes of alcohol (100%), 3 min each and cleared through 3 changes of xylene (Fisher Chemicals), 3 min each. Slides were mounted using Permount solution (Fisher).
For whole-mounts staining of large intestines, tissue fragments (1–2cm) were fixed in freshly prepared 4% paraformaldehyde (PFA) in PBS for 6–8h and incubated in 0.5% (wt/vol) saponin, 2% fetal bovine serum (FBS) (vol/vol), and 0.09% (wt/vol) azide in PBS for at least 18 h at 4°C. The same buffer was used for subsequent incubations with fluorescently labeled Phalloidin (Alexa Fluor 647) and YOYO-1 iodide (491/509) overnight at 4°C followed by 2h incubation at 37°C. Samples were washed with PBS and analyzed by microscopy.
TUNEL assay/Immunofluorescence staining
Frozen 5μm aortic roots were fixing for 10 min in acetone (Sigma-Aldrich) at RT and placed into 1% paraformaldehyde in 100mM sodium phosphate containing 60mM lysine and 7mmM sodium periodate (Sigma-Aldrich) pH 7.4 on ice. Then slides were permeabilized with freshly prepared 0.1% Triton X-100, 0.1% sodium citrate solution and incubated with TUNEL reaction mixture for 60 minutes at 37°C in the dark. The sections were i ncubated with avidin/biotin blocking kit (Vector laboratories) followed by incubation in a 5% normal goat serum and 1% BSA (Sigma) in PBS. Aortic root sections were stained overnight at 4°C with primary antibodies: rat anti-CD11bFITC (M1/70, BD Bioscience) followed by staining with secondary antibodies for 1 h at RT: goat anti-FITC Alexa Fluor 488 (Molecular Probes).
Gene expression analysis
The tissues (aorta, intestine or spleen) were homogenized with RNAse/DNase free 2.8mm Ceramic Beads using Omni Bead Ruptor 24 in PureZOL RNA Isolation Reagent (Bio-Rad Laboratories) followed by RNA isolation using Aurum Total RNA Fatty and Fibrous Tissue kit (Bio-Rad Laboratories) according to manufacturer’s protocol. First strand cDNA was synthetized using the iScript Reverse Transcription Supermix (Bio-Rad Laboratories). Gene expression was analyzed by SYBR green real-time polymerase chain reaction (Bio-Rad Laboratories) using primers for RpL32, Spp1, Il1b, Tnf, Ifng, Il17a, Il22, Ccl2, Ccl5, Cxcl1, Reg3b, Reg3g, Muc2, Cd36, Itgam, Il12a, Il12b, Mrc1, Chi3l1, Chi3l3, Retnla, Abca1, Abcg1 (mouseprimerdepot.nci.nih.gov). Gene expression was normalized to RpL32 expression. Data were analyzed using Prism statistical software (GraphPad).
Microbiome analysis
16S rRNA sequencing:
To analyze 16S rRNA of gut microbiota, bacterial DNA was isolated from a fecal microbiome of WT, Il23−/− and Il22−/− bone marrow transplanted Ldlr−/− mice. The stools were homogenized with RNAse/DNase free 2.8mm Ceramic Beads using Omni Bead Ruptor 24 in ASL buffer (stool lysis buffer) (Qiagen) followed by DNA isolation using QIAamp DNA stool Mini Kit (Qiagen) according to manufacturer’s protocol. QPCR was carried out using universal or bacterial strain-specific primers for 16S rRNA genes. The V4 region of the 16S rDNA gene using two round amplification PCR using Phusion High-Fidelity DNA-polymerase (NEB) with 100ng of input DNA. First round PCR (15 cycles):
515F TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG
806R GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC
Second round PCR (10 cycles) with i5 and i7 Illumina barcode sequences:
AATGATACGGCGACCACCGAGATCTACAC-i5-TCGTCGGCAGCGTC
CAAGCAGAAGACGGCATACGAGAT-i7-GTCTCGTGGGCTCGG
Amplicones were purified between and after the PCR reactions with Agencourt AMPure XP (Beckman Coulter, Inc.), libraries were quantified using KAPA Library Quantification Kit for Illumina Platforms (Kapa Biosystems) and size was estimated using Agilent Tapestation 4200 (Agilent). Libraries were pooled at equimolar concentration using Epmotion 5075 tmx automatic liquid handling machine (Eppendorf) and sequenced with 5% PhiX library added to the pool on Illumina MiSeq machine according to manufacture instructions using v3 kit (600 cycles). Fastq files were generated on Basespace (Illumina). On average 100,000 reads were obtained per sample, and after quality control filtering; on average 90,000 reads per sample were processed further. Chimeric sequences were filtered out of the FASTQ files containing the 16S rRNA gene sequences using the USEARCH (version 8.1.) utility’s UCHIME implementation and the ‘gold’ database (version microbiomeutil-r20110519). The reads, thus filtered, were then binned into operational taxonomic units (OTUs) at 97% similarity using USEARCH’s cluster_otus command. QIIME (1.9.1) scripts were used to classify and align the obtained OTUs (Namasivayam et al., 2017). The assign_taxonomy.py script was used to assign taxonomy using the default RDP method (Wang et al., 2007) and GreenGenes database (DeSantis et al., 2006). The statistical differences between the groups were estimated with non-parametric Mann-Whittney test and pvalues were corrected for multiple tests using q-value 0.05 as a cutoff and data was visualized using heatmaps and PCA plots (Partek 6.6).
Metagenomic whole genome shotgun (WGS) sequencing
Metagenomic sequencing provides species-level resolution of bacteria, and depending on sequence depth, can quantify the near-complete genomic content of the collection of microbes in a particular sample, referred to as the sample’s metagenome.
Whole genome shotgun (WGS) sequencing utilized the same extracted bacterial genomic DNA used for 16S rRNA gene compositional analysis and was done at Microbiome Core at NCI. Briefly, metagenomic DNA obtained from 16 samples was used to build paired-end shotgun sequencing libraries with the Illumina Nextera FLEX kit. Libraries were sequenced on the Illumina NextSeq 500 platform yielding 2 × 75 bp reads totaling between 2.9 and 4.9 Gbp of raw data. For each sample, raw reads were adapter trimmed and quality clipped using Trimmomatic v.0.36 (Bolger et al., 2014). Trimmed reads were then aligned against the Mus musculus complete genome using bowtie v.2–2.3.4. Reads which did not align to the mouse genome were considered as pertaining to the microbiome and were used as input for read assembly using MEGAHIT v1.1.1 (Li et al., 2015). The mean assembly rate for samples was 69% ± 13% of total reads being assembled into contigs of at least 500 bp. Contigs resulting from assembly were taxonomically classified by k-mer spectrum with kraken v.1.1 (Wood and Salzberg, 2014) using a custom built k-mer database comprising of all complete plus draft genomes present in NCBI’s GenBank of bacteria, archaea, viruses, fungi, protozoa, plus the genomes of Homo sapiens and Mus musculus. Contigs identified as being Mus musculus were discarded. Contigs were annotated using prokka v.1.12 (Seemann, 2014) and the predicted metaproteome was functionally annotated using interproscan v.5.29–68.0 (Hunter et al., 2009). The trimmed reads used as input for assembly were then aligned against the contigs using bowtie2 to gauge sequencing depth for each contig. Reads, which did not align to any contig (unassembled reads), were collected and themselves taxonomically classified by kraken using the aforementioned custom-built k-mer database. The relative abundance for each taxon was calculated as being the relative proportion of bases attributed to that taxon, coming from either unassembled reads or reads built into contigs, out of the total amount of bases sequenced for that sample. Relative abundances were expressed as parts per million.
The relative abundance of functions (taxonomy independent) was calculated in a similar manner, but considering only bases assembled into contigs, as each protein was predicted ab initio from the contigs. The number of bases sequenced for each protein-coding gene was measured from the contig sequencing depth. Because all the proteins were functionally characterized by Interproscan as to their functional domain signatures, the relative abundance of each functional protein signature within a sample was computed as being the relative proportion (expressed in parts per million) of the sum of the sequencing depths of all protein coding genes bearing that signature out of the sum of the sequencing depths of all the predicted proteins within the sample. It can thus be interpreted as the “dose” of a functional signature within the sample, independent of taxonomy.
Additional factor accounting for cage effect was added in the statistical model testing for unique and common effect of IL-22 and IL-23 deficiency in atherosclerotic mice.
Serum analysis of bacterial endotoxin (LPS)
Blood samples were drawn from WT, Il23−/− or Il22−/− transplanted Ldlr−/− mice and serums were collected via centrifugation for 10 min. Serum endotoxin level was determined using Pierce LAL Chromogenic Endotoxin Quantification Kit (Thermo Scientific) according to manufacture protocol. The results were analyzed using spectrophotometer.
TMAO analysis
TMAO levels were quantified by stable isotope dilution liquid chromatography with on line electrospray ionization tandem mass spectrometry on a Shimadzu 8050 triple quadrupole mass spectrometer interfaced with UHPLC using d9(trimethyl)-TMAO as internal standard, as previously described (Wang et al., 2014b).
Metabolomics
Mass spectrometry analysis was performed by the Proteomics and Metabolomics Facility at the Wistar Institute. Polar metabolites were extracted from serum using 80% methanol, and targeted quantitation of metabolites was performed on a SCIEX 5500 QTRAP triple quadrupole mass spectrometer equipped with a Turbo V source and coupled to a Shimadzu Nexera UHPLC system. Five microliters of each sample were injected onto a ZIC-pHILIC 2.1-mm i.d × 150 mm column (EMD Millipore). Buffer A was 20 mM ammonium carbonate, 0.1% ammonium hydroxide; and buffer B was acetonitrile. The chromatographic gradient was run at a flow rate of 0.150 ml/min for 80–20% B over 20 min, 20–80% B over 0.5 min, and hold at 80% B for 7.5 min. Metabolites were targeted in positive and negative ion modes for a total of 163 selected reaction monitoring transitions. Electrospray ionization voltage was +5500 V in positive ion mode and −4500 V in negative ion mode. A minimum of 8 data points was acquired per detected metabolite. Peak areas from the total ion current for each metabolite-selected reaction monitoring transition were integrated using the MultiQuant v3.0.2 software (SCIEX).
Metabolome detection intensity levels were quantile normalized and log2-transformed. Significance of changes were tested using Limma package (Ritchie et al., 2015) version 3.36.1 combining mice genders and separately and results passing p<0.05 threshold were called significant. Additional factor accounting for cage effect was added in the statistical model testing for unique and common effect of IL-22 and IL-23 deficiency in atherosclerotic mice. Results related to TMAO pathway were reported as fold changes with significant effects highlighted.
LPS and TMAO supplementation
Beginning at 6 weeks of age, female and male Ldlr−/− were fed with a WD for 6 weeks and then injected i.p. with LPS (50μg per injection), TMAO (250 μl of 100μM solution per injection) or both metabolites together every 3 days for additional 3 weeks of WD feeding. After that, changes in atherosclerosis progression and inflammatory gene expression were analyzed.
For the ex vivo experiments explanted aortas from C57BL/6 mice were stimulated in serum free DMEM media with LPS (100ng/ml), TMAO (200ng/ml) or both for 8h at 37°C in CO 2 incubator. For acute LPS and TMAO exposure studies, C57BL/6 female and male mice were injected with LPS (50μg) and TMAO (250 μl of 100μM solution per injection) and blood was collected after 4 hours following by immunomagnetic isolation of Ly6C positive monocytes using Easy Sep FITC Positive Selection Kit (StemCell Technologies) according to the manufacture protocol. Sorted cells were lysed in RLT buffer following with RNA isolation and Q-RT-PCR analysis.
Scanning Electron Microscopy
Proximal colon and terminal ileum were collected from WT ➔Ldlr−/−, Il23−/− ➔Ldlr−/− and Il22−/− ➔Ldlr−/− mice fed with WD for 16 weeks for SEM imaging. Intestinal samples were fixed in 2.5% glutaraldehyde in 0.1 M PBS (pH 7.4) for 10 min. Tissue samples were washed thoroughly in 0.1 M PBS for 15 min. Further, tissues were fixed in 1% Osmium tetraoxide (OsO4) in 0.1 M PBS for 60 min. Samples were then dehydrated with different concentration of ethanol 30%, 50%, 70%, 90% for 15 min and then samples were critical point dried. Finally, samples were sputter coated with gold. Samples were visualized using Field Emission Scanning Electron Microscope (JEOL JSM 6335F) at the magnification of 550X and 1000X.
Quantification and Statistical analysis
Data were analyzed using Prism software (GraphPad). Student’s 2-tailed T-test and Wilcoxon signed-rank test was used to compare fold induction of gene expression by real-time PCR.
ANOVA test was used for multiple comparisons.
Microbiota composition was analyzed and visualized using Partek, Matlab and R packages. Standard statistical tests, such as t-test, ANOVA, PERMANOVA, PCA, t-SNE and others were performed and visualized using scatter plots and heatmaps. Picrust analysis was performed on biome file. Pathways outputs were analyzed using Partek and R. Data are expressed as mean ± SEM. *p<0.05, ** p <0.01, *** p <0.001. P value <0.05 was considered significant.
Supplementary Material
Highlights.
IL-23 and IL-22 cytokines restrict atherosclerosis development
IL-23 and IL-22 control intestinal barrier and pro-atherogenic bacteria expansion
Bacterial metabolites activate myeloid cells and drive atherosclerosis
Acknowledgements
We thank Laboratory animal, Flow cytometry, Cell culture and Microscopy facilities at FCCC and Genentech Inc for providing Il23p19−/− and Il22−/− mice and rIL-22-Ig. We thank Dr. Klaus Ley (La Jolla Institute) for help at early stages of this work and Drs. Daniel Mucida (Rockefeller University), Siddharth Balachandran (FCCC) and Rachel E. Locke for critical comments. This work was supported by Pew Scholar Award to S.I.G., NIH grants NCI P30 Cancer Center Grant to FCCC; R21 CA202396, R56 HL133669, R01 HL133669 to E.K.K. and R01HL103866 to S.L.H. S.L.H. also reports being partially supported by a Leducq Foundation Award.
Footnotes
Declaration of interest: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbas A, Gregersen I, Holm S, Daissormont I, Bjerkeli V, Krohg-Sorensen K, Skagen KR, Dahl TB, Russell D, Almas T, et al. (2015). Interleukin 23 levels are increased in carotid atherosclerosis: possible role for the interleukin 23/interleukin 17 axis. Stroke 46, 793–799. [DOI] [PubMed] [Google Scholar]
- Ahern PP, Schiering C, Buonocore S, McGeachy MJ, Cua DJ, Maloy KJ, and Powrie F (2010). Interleukin-23 drives intestinal inflammation through direct activity on T cells. Immunity 33, 279–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ait-Oufella H, Taleb S, Mallat Z, and Tedgui A (2011). Recent advances on the role of cytokines in atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology 31, 969–979. [DOI] [PubMed] [Google Scholar]
- Behnsen J, Jellbauer S, Wong CP, Edwards RA, George MD, Ouyang W, and Raffatellu M (2014). The cytokine IL-22 promotes pathogen colonization by suppressing related commensal bacteria. Immunity 40, 262–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkaid Y, and Hand TW (2014). Role of the microbiota in immunity and inflammation. Cell 157, 121–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brusca SB, Abramson SB, and Scher JU (2014). Microbiome and mucosal inflammation as extra-articular triggers for rheumatoid arthritis and autoimmunity. Current opinion in rheumatology 26, 101–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caligiuri G, Rudling M, Ollivier V, Jacob MP, Michel JB, Hansson GK, and Nicoletti A (2003). Interleukin-10 deficiency increases atherosclerosis, thrombosis, and low-density lipoproteins in apolipoprotein E knockout mice. Mol Med 9, 10–17. [PMC free article] [PubMed] [Google Scholar]
- Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, et al. (2003). Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 421, 744–748. [DOI] [PubMed] [Google Scholar]
- Desai MS, Seekatz AM, Koropatkin NM, Kamada N, Hickey CA, Wolter M, Pudlo NA, Kitamoto S, Terrapon N, Muller A, et al. (2016). A Dietary Fiber-Deprived Gut Microbiota Degrades the Colonic Mucus Barrier and Enhances Pathogen Susceptibility. Cell 167, 1339–1353 e1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, and Andersen GL (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72, 5069–5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, Peaper DR, Bertin J, Eisenbarth SC, Gordon JI, and Flavell RA (2011). NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145, 745–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelbertsen D, Depuydt MAC, Verwilligen RAF, Rattik S, Levinsohn E, Edsfeldt A, Kuperwaser F, Jarolim P, and Lichtman AH (2018). IL-23R Deficiency Does Not Impact Atherosclerotic Plaque Development in Mice. Journal of the American Heart Association 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J, Bonder MJ, Cenit MC, Tigchelaar EF, Maatman A, Dekens JA, Brandsma E, Marczynska J, Imhann F, Weersma RK, et al. (2015). The Gut Microbiome Contributes to a Substantial Proportion of the Variation in Blood Lipids. Circulation research 117, 817–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffen SL, Jain R, Garg AV, and Cua DJ (2014). The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol 14, 585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galkina E, and Ley K (2009). Immune and inflammatory mechanisms of atherosclerosis. Annu Rev Immunol 27, 165–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman AL, and Gordon JI (2010). Our unindicted coconspirators: human metabolism from a microbial perspective. Cell metabolism 12, 111–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory JC, Buffa JA, Org E, Wang Z, Levison BS, Zhu W, Wagner MA, Bennett BJ, Li L, DiDonato JA, et al. (2015). Transmission of atherosclerosis susceptibility with gut microbial transplantation. J Biol Chem 290, 5647–5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, Taniguchi K, Yu GY, Osterreicher CH, Hung KE, et al. (2012). Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 491, 254–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Kitamoto S, Wang H, and Boisvert WA (2010). Interleukin-10 overexpression in macrophages suppresses atherosclerosis in hyperlipidemic mice. FASEB J 24, 2869–2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson GK, and Hermansson A (2011). The immune system in atherosclerosis. Nature immunology 12, 204–212. [DOI] [PubMed] [Google Scholar]
- Hornemann T, and Worgall TS (2013). Sphingolipids and atherosclerosis. Atherosclerosis 226, 16–28. [DOI] [PubMed] [Google Scholar]
- Hue S, Ahern P, Buonocore S, Kullberg MC, Cua DJ, McKenzie BS, Powrie F, and Maloy KJ (2006). Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J Exp Med 203, 2473–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter S, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, Bork P, Das U, Daugherty L, Duquenne L, et al. (2009). InterPro: the integrative protein signature database. Nucleic acids research 37, D211–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isoda K, Kamezawa Y, Ayaori M, Kusuhara M, Tada N, and Ohsuzu F (2003). Osteopontin transgenic mice fed a high-cholesterol diet develop early fatty-streak lesions. Circulation 107, 679–681. [DOI] [PubMed] [Google Scholar]
- Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, et al. (2009). Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson AL, and Backhed F (2017). Role of gut microbiota in atherosclerosis. Nat Rev Cardiol 14, 79–87. [DOI] [PubMed] [Google Scholar]
- Kahles F, Findeisen HM, and Bruemmer D (2014). Osteopontin: A novel regulator at the cross roads of inflammation, obesity and diabetes. Mol Metab 3, 384–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada N, Seo SU, Chen GY, and Nunez G (2013). Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol 13, 321–335. [DOI] [PubMed] [Google Scholar]
- Kelly TN, Bazzano LA, Ajami NJ, He H, Zhao J, Petrosino JF, Correa A, and He J (2016). Gut Microbiome Associates With Lifetime Cardiovascular Disease Risk Profile Among Bogalusa Heart Study Participants. Circulation research 119, 956–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khojasteh-Fard M, Abolhalaj M, Amiri P, Zaki M, Taheri Z, Qorbani M, Bazzaz JT, and Amoli MM (2012). IL-23 gene expression in PBMCs of patients with coronary artery disease. Dis Markers 33, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosravi A, and Mazmanian SK (2013). Disruption of the gut microbiome as a risk factor for microbial infections. Curr Opin Microbiol 16, 221–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Liu CM, Luo R, Sadakane K, and Lam TW (2015). MEGAHIT: an ultra-fast singlenode solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. [DOI] [PubMed] [Google Scholar]
- Matsui Y, Rittling SR, Okamoto H, Inobe M, Jia N, Shimizu T, Akino M, Sugawara T, Morimoto J, Kimura C, et al. (2003). Osteopontin deficiency attenuates atherosclerosis in female apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 23, 1029–1034. [DOI] [PubMed] [Google Scholar]
- Namasivayam S, Maiga M, Yuan W, Thovarai V, Costa DL, Mittereder LR, Wipperman MF, Glickman MS, Dzutsev A, Trinchieri G, and Sher A (2017). Longitudinal profiling reveals a persistent intestinal dysbiosis triggered by conventional anti-tuberculosis therapy. Microbiome 5, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postler TS, and Ghosh S (2017). Understanding the Holobiont: How Microbial Metabolites Affect Human Health and Shape the Immune System. Cell metabolism 26, 110–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattik S, Hultman K, Rauch U, Soderberg I, Sundius L, Ljungcrantz I, Hultgardh-Nilsson A, Wigren M, Bjorkbacka H, Fredrikson GN, and Nilsson J (2015). IL-22 affects smooth muscle cell phenotype and plaque formation in apolipoprotein E knockout mice. Atherosclerosis 242, 506–514. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, et al. (2017). Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med 377, 1119–1131. [DOI] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic acids research 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savvatis K, Pappritz K, Becher PM, Lindner D, Zietsch C, Volk HD, Westermann D, Schultheiss HP, and Tschope C (2014). Interleukin-23 deficiency leads to impaired wound healing and adverse prognosis after myocardial infarction. Circulation. Heart failure 7, 161–171. [DOI] [PubMed] [Google Scholar]
- Schroeder BO, and Backhed F (2016). Signals from the gut microbiota to distant organs in physiology and disease. Nature medicine 22, 1079–1089. [DOI] [PubMed] [Google Scholar]
- Seemann T (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 20682069. [DOI] [PubMed] [Google Scholar]
- Subramanian M, Thorp E, and Tabas I (2015). Identification of a non-growth factor role for GM-CSF in advanced atherosclerosis: promotion of macrophage apoptosis and plaque necrosis through IL-23 signaling. Circulation research 116, e13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto K, Ogawa A, Mizoguchi E, Shimomura Y, Andoh A, Bhan AK, Blumberg RS, Xavier RJ, and Mizoguchi A (2008). IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest 118, 534–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tailford LE, Crost EH, Kavanaugh D, and Juge N (2015). Mucin glycan foraging in the human gut microbiome. Frontiers in genetics 6, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taleb S, Tedgui A, and Mallat Z (2010). Interleukin-17: friend or foe in atherosclerosis? Curr Opin Lipidol 21, 404–408. [DOI] [PubMed] [Google Scholar]
- Wang Q, Garrity GM, Tiedje JM, and Cole JR (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73, 5261–5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Ota N, Manzanillo P, Kates L, Zavala-Solorio J, Eidenschenk C, Zhang J, Lesch J, Lee WP, Ross J, et al. (2014a). Interleukin-22 alleviates metabolic disorders and restores mucosal immunity in diabetes. Nature 514, 237–241. [DOI] [PubMed] [Google Scholar]
- Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM, et al. (2011). Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 472, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Levison BS, Hazen JE, Donahue L, Li XM, and Hazen SL (2014b). Measurement of trimethylamine-N-oxide by stable isotope dilution liquid chromatography tandem mass spectrometry. Anal Biochem 455, 35–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Roberts AB, Buffa JA, Levison BS, Zhu W, Org E, Gu X, Huang Y, Zamanian-Daryoush M, Culley MK, et al. (2015). Non-lethal Inhibition of Gut Microbial Trimethylamine Production for the Treatment of Atherosclerosis. Cell 163, 1585–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiekowski MT, Leach MW, Evans EW, Sullivan L, Chen SC, Vassileva G, Bazan JF, Gorman DM, Kastelein RA, Narula S, and Lira SA (2001). Ubiquitous transgenic expression of the IL-23 subunit p19 induces multiorgan inflammation, runting, infertility, and premature death. Journal of immunology 166, 7563–7570. [DOI] [PubMed] [Google Scholar]
- Wood DE, and Salzberg SL (2014). Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome biology 15, R46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Q, Gu Y, Li X, Yang W, Jia L, Chen C, Han X, Huang Y, Zhao L, Li P, et al. (2017). Alterations of the Gut Microbiome in Hypertension. Frontiers in cellular and infection microbiology 7, 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, Abbas AR, Modrusan Z, Ghilardi N, de Sauvage FJ, and Ouyang W (2008). Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nature medicine 14, 282–289. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.