

Abstract
The efficacy of the adaptive immune response declines dramatically with age, but the cell-intrinsic mechanisms driving immune aging in humans remain poorly understood. Immune aging is characterized by a loss of self-renewing naïve cells and the accumulation of differentiated but dysfunctional cells within the CD8 T cell compartment. Using ATAC-seq, we inferred the transcription factor binding activities correlated with naive and central and effector memory CD8 T cell states in young adults. Integrating our results with RNA-seq, we identified transcription networks associated with CD8 T cell differentiation, with prominent roles implicated for BATF, ETS1, Eomes, and Sp1. Extending our analysis to aged humans, we found that the differences between the memory and naive subsets were largely preserved across age, but that naive and central memory cells from older individuals exhibited a shift toward more differentiated patterns of chromatin openness. Additionally, aged naive cells displayed a loss in chromatin accessibility at gene promoters, largely associated with a decrease in NRF1 binding. This shift was implicated in a marked drop-off in the ability of the aged naive cells to transcribe respiratory chain genes, which may explain the reduced capacity of oxidative phosphorylation in older naïve cells. Our findings identify BATF- and NRF1-driven gene regulation as potential targets for delaying CD8 T cell aging and restoring function.
Introduction
Aging is a ubiquitous biological process that drastically alters physiological function. Correspondingly, immune competency erodes with age, leading to higher incidences of malignancies and increased morbidity and mortality from infections (1, 2). Much of this decline has been attributed to a breakdown in T-cell homeostasis, degrading the competence of the central mediator of adaptive immunity (3, 4). Absent thymic production, T cell renewal in humans is entirely driven by the proliferation of peripheral naïve T cells (5). This homeostatic proliferation is similar across naïve CD4 and CD8 T cells, independent of age (6), and maintains a diverse naïve T cell repertoire in aged individuals (7). However, while the CD4 T cell compartment is stable, age drastically affects the distribution of CD8 subsets, causing a diminution of naïve and central memory cells and an expansion of effector cells (8). Additionally, naïve T cells develop functional defects with age that may relate to replicative stress induced by homeostatic turnover (9). Characterizing these cell-intrinsic drivers of T cell aging and their relationship to differentiation is critical in identifying and combating immune aging.
Changes to the epigenetic landscape are hallmarks of both aging and differentiation, and encompass chromatin remodeling, histone modifications, and DNA methylation (10, 11). Significant revisions to the epigenetic landscape are required for naïve T cells to lose their stem cell-like attributes and develop into memory cells that rapidly respond to antigenic stimulation (12). Limited genome-wide epigenetic studies have been performed on human CD8 T cell subsets; they have not comprehensively profiled chromatin accessibility in naive, effector, and central memory compartments, nor have they examined the baseline regulatory landscape for age-related dysfunction (13–15).
Here, we report the first genome-wide maps of chromatin accessibility in purified CD8 T cell subsets from age-segregated human donors. These data enabled us to define the changes across differentiation by comparing naïve, effector, and central memory cells, and then to determine how those regulatory programs, as well as the subsets’ baseline chromatin landscapes, differ with age. Our results describe immune aging of CD8 naïve and central memory T cells as a shift toward a more differentiated state, coupled with an erosion of chromatin accessibility at key promoters, in part mediated by a loss of NRF1 binding.
Results
Genome-wide mapping of chromatin accessibility in CD8 T cell subsets
Using the assay for transposase-accessible chromatin (ATAC-seq) (16, 17), we generated genome-wide maps of chromatin accessibility in three human CD8 T cell subsets, purified by sorting peripheral blood mononuclear cells (PBMC): naïve (CD28hiCD45RAhiCD62Lhi), central memory (CD45RAloCD62Lhi), and effector memory (CD45RAloCD62Llo; Figures 1A and S1). The PBMC were isolated from ten healthy individuals, comprising five donors under age 35 (“young;” 2 males and 3 females) and five over age 60 (“old;” 3 males and 2 females). We excluded terminally differentiated CD28loCD45RAhi effector T cells, which mostly contain oligoclonally expanded herpes-virus-specific cells, to focus on age-related chromatin changes rather than changes induced by chronic viral stimulation. To support the functional relevance of the chromatin openness patterns we identified, we also performed RNA-seq in each of the T cell subsets.
Fig. 1. ATAC-seq open chromatin maps capture characteristic signatures that correlate with phenotypically defined CD8 T cell subsets.

(A) T cells were sorted into naïve (CD28hiCD45RAhiCD62Lhi), central memory (CM; CD45RAloCD62Lhi), and effector memory (EM; CD45RAloCD62Llo) CD8 T cell subsets, as shown in Fig. S1. Contour plots show purity of sorted populations.
(B) ATAC-seq signal tracks in naive, CM, and EM subsets at selected functionally relevant genes (CD8, CD4, IFNG and GZMB). Small boxes in gene diagrams (bottom) represent exons. Each subset’s count is aggregated across the ten constituent donors. The y-axes are in units of reads per million reads in peaks.
(C) Proportion of chromatin states in naive CD8 T cells, genome-wide (left), and for ATAC-seq peaks (right). Chromatin states were called by the Roadmap Epigenomics Project using ChromHMM, and we aggregated the state calls over functionally related subclasses. Percentages are as fractions of base pairs.
(D) Heat map of Spearman correlation coefficients of openness between naïve (Na), CM, and EM samples. Dendrograms indicate results from consensus clustering.
Sequencing read density at phenotype-defining genes confirmed successful transposition in each subset, with accessible chromatin at the CD8A locus and minimal accessibility at the CD4 locus (Fig. 1B). IFNG and GZMB, which are known to be more rapidly inducible in memory T cells after activation, were more open in central (CM) and effector memory (EM) than naïve cells.
Across all three subsets, we identified a total of 64,089 distinct chromatin accessibility peaks. Using predictions of chromatin states in naïve CD8 T cells from the Roadmap Epigenomics Project (18), we observed that the peaks were concentrated at promoters and enhancers, and depleted at heterochromatic regions (Fig. 1C). Hierarchical clustering of the peaks segregated the three T cell subsets (Fig. 1D), with the two memory populations clustering more closely with each other than with the naïve population. To mitigate the impact of donor heterogeneity, we excluded sex chromosomes, which have been shown to be a major site of inter-individual variation in chromatin accessibility (19).
To uncover the chromatin accessibility landscapes underlying naïve and memory cell identity in the young, we identified loci of differential accessibility between each pair of subsets using DESeq2 (20). We observed greater numbers of differentially open loci between naïve and memory subsets than between the two memory subsets (Fig. 2A); comparing young naive to young EM yielded 18,489 differentially accessible peaks (28.8% of those tested), while comparing young CM to young EM resulted in only 914 (1.4%). To determine which molecular pathways were subject to regulation, we performed k-means clustering of the differentially open peaks (Fig. 2B) and used GREAT (21) to calculate gene ontology (GO) enrichment for each cluster, separately for peaks for which openness was positively versus negatively correlated with expression of the nearest gene. We found that each cluster was enriched for GO terms reflecting the underlying T cell biology (Figures 2B and S2A). The accessibility of the peaks most open in CM and closed in naive (cluster 1; red) was positively correlated with adaptive immune response pathways, particularly those regulating leukocyte cell-to-cell adhesion. Accessibility of the peaks most open in EM and closed in naive (cluster 2; blue) was correlated with cytokine production, concordant with the increased effector cytokine production of EM cells. Moreover, peaks in this cluster were associated with the up-regulation of genes that repress cell activation, and the down-regulation of genes that facilitate activation, suggesting the induction of negative regulatory pathways in effector cells. This pattern held for genes regulating proliferation, consistent with the loss of proliferative potential following differentiation. Accessibility of the peaks more open in naive (cluster 3; green) was associated with the up-regulation of T cell proliferation, T cell receptor signaling, and V(D)J recombination, representing features of T cell biology unique to naïve cells and their progenitors. Thus, changes in chromatin accessibility across CD8 T cell differentiation were associated with the regulatory pathways that define the T cell subsets.
Figure 2. Differentially open chromatin loci form distinct clusters and neighbor immunologically relevant genes.
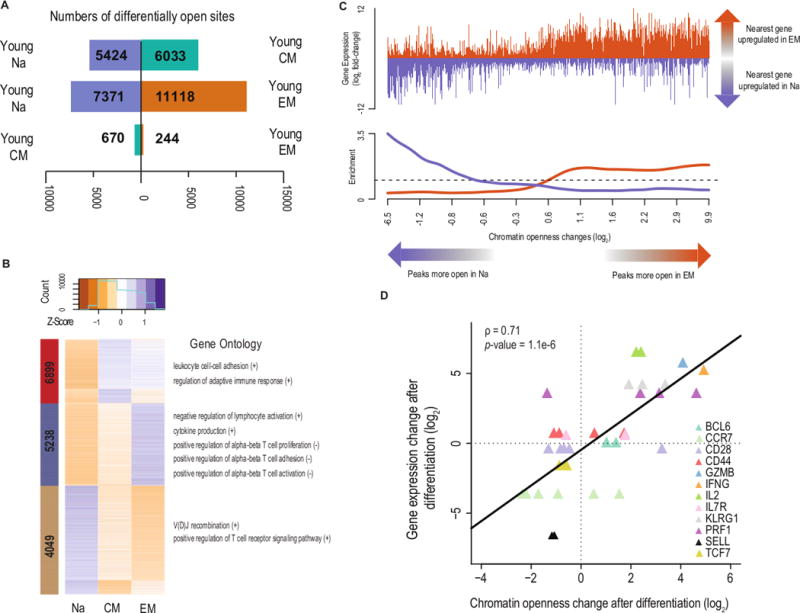
(A) Numbers of peaks significantly changing in openness after differentiation. Directions of bars indicate in which subset the peaks were more open.
(B) k-means clustering of peaks differentially open across differentiation. Peaks are in rows and colors represent z-scores of openness. The sidebar colors label the three clusters, with the inset numbers giving the numbers of peaks in each. Adjacent are results from GO analysis of each cluster, with peaks separately analyzed based on whether the relative chromatin openness was positively (+) or negatively (-) correlated with the expression of the nearest expressed gene.
(C) Enrichment for gene expression changing coordinately with chromatin openness changes. The top panel shows the changes of each peak’s nearest gene, with genes increasing in expression with differentiation indicated in orange and genes decreasing in expression shown in purple. The bottom panel is a smoothed track of the fold-changes, divided by the global fold-change average.
(D) Correlation of chromatin openness changes and gene expression changes of selected canonical differentiation-associated genes. One point is plotted for each peak for which the nearest gene was one of the 12 differentiation-associated genes depicted. Black line indicates the least-squares fit.
We next tested whether changes in chromatin openness were directly correlated with changes in gene expression. We found that peaks more open after differentiation also neighbored genes more expressed in the differentiated cells. Peaks that were closed after differentiation likewise neighbored genes less expressed after differentiation (Fig. 2C). Furthermore, when the openness changes were large, neighboring genes were more likely to change in expression (GSEA p = 0.03), and the expression changes became more extreme (Fig. S2B). This finding held both for canonical naïve and effector genes (correlation between accessibility and expression changes = 0.71, p = 1.1 * 10−6; Fig. 2D) and overall (p < 2.2 * 10−16, chi-squared test; Fig. S2C). These observations indicate that chromatin accessibility changes across CD8 T cell differentiation are associated with the regulation of functionally relevant pathways and genes.
Maintenance of naïve and memory T cell states is associated with distinct transcription factor families
TF binding can instigate chromatin remodeling and drive differentiation (22, 23). To identify TFs putatively associated with differentiation in T cells, we calculated the enrichment of TF binding motifs in the loci that changed in accessibility after differentiation. At peaks more open in EM than naive (Fig. 3A), we found motifs for several members of the bZIP, T-box, and RUNT TF families among the most significantly enriched (Table S1). Our top hits encompassed known regulators of T cell differentiation and development, such as Eomes and TBX21 (T-bet) (FDR < 10−66; (24)). The TF family with the greatest representation was bZIP, which includes Jun-AP1 and BATF, both critical to T cell differentiation (25). These TFs were also more expressed in EM than naive. The only bZIP TF more expressed in naive cells than effector cells was Bach2, which is known to suppress effector-related genes to maintain a naive state (26); this TF also shares strong motif similarity with other bZIP TFs. Some TFs known to be overexpressed in EM, such as PRDM1 (27, 28), showed smaller, yet significant, binding motif enrichment. The motif enrichment in peaks more accessible in EM than in CM was modest (Table S1), as expected due to their similarity in accessibility patterns. We did observe minor enrichment for the binding motifs of T-bet and Eomes, which are known to be more expressed in effector than central memory populations (29). Overall, the finding of known regulators confirmed that our inference of TF binding is concordant with current models of human CD8 T cell differentiation.
Fig. 3. Chromatin accessibility peaks are associated with distinct TF families in differentiated CD8 T cell subsets.
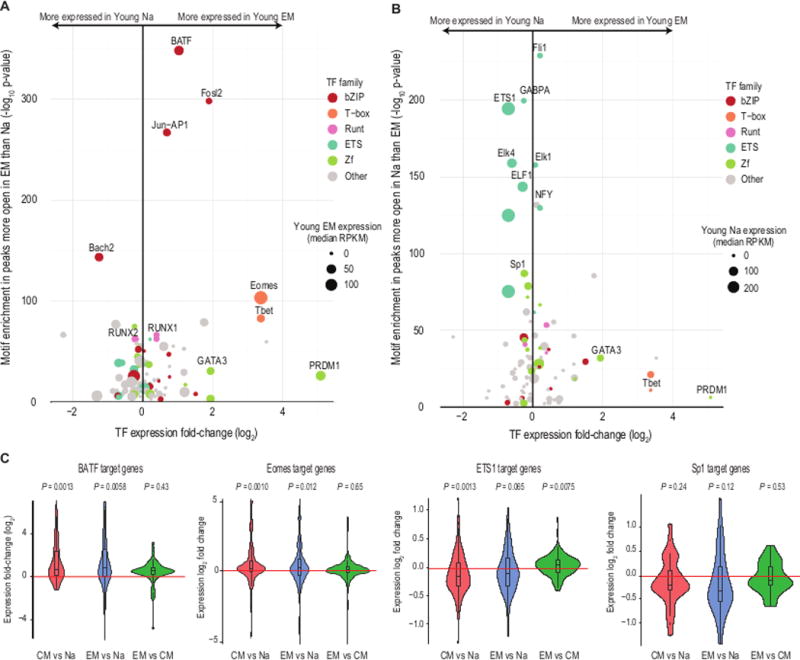
(A) Expression of TFs with binding motifs enriched in loci more accessible following differentiation. The x-axis indicates TF expression fold-change following differentiation from naïve (Na) to EM, and the y-axis indicates the –log(p-value) of TF binding motif enrichment in loci that are more open after differentiation. Dot size indicates median TF expression in the EM, and dot color gives the TF family to which the TF belongs, based on the DNA-binding motif. The statistical significance of the enrichment of all TF binding motifs in differentially accessible peaks is given in Table S1.
(B) As in (A), for TFs with binding motifs enriched in loci less accessible following differentiation. Here, dot size indicates TF expression level in the Na.
(C) Violin plots and boxplots of the log2(fold-changes) of expression levels of gene targets of BATF, Eomes, ETS1, and Sp1 between the T cell subset comparisons. p-values give results of Wilcoxon rank-sum tests assuming a median log2(fold-change) of 0 (red line). Target sets are drawn from published knockdown experiments in human-derived cell lines (25, 32–35).
Peaks that closed following differentiation were enriched for motifs recognized by the ETS and zinc finger (Zf) families of TFs (Fig. 3B), and most ETS and Zf TFs were more expressed before differentiation. ETS TFs have been shown to be required for normal T cell development, survival, and activation (30, 31), but they have not previously been reported to be central to the epigenetic maintenance of the naïve T cell state. Taken together, the motif enrichment, gene expression levels, and gene expression changes nominate BATF and Eomes as key factors in the epigenetic changes in CD8 T cell differentiation, and ETS1 and Sp1 in maintaining the chromatin accessibility landscape of naïve cells.
We hypothesized that greater TF expression plus greater chromatin openness at the TFs’ binding motifs would lead to greater expression of the TFs’ target genes. We evaluated this hypothesis using published TF target sets from knockdown experiments in human-derived cell lines (25, 32). We found that the majority of BATF targets increased in expression following differentiation (p = 0.001, young CM versus naive, and p = 0.006, young EM versus naive, Wilcoxon rank-sum tests; Fig. 3C). The trend was similar for Eomes targets (p = 0.001, young CM versus naive, and p = 0.01, young EM versus naive). Expression of BATF and Eomes targets did not differ uniformly between the two memory populations (p = 0.43 and 0.65, respectively), consistent with the high degree of epigenetic similarity between the memory populations.
In the reciprocal analysis, we evaluated ETS1 and Sp1 target genes (33–35). We found that the majority of ETS1 target genes decreased in expression following differentiation (p = 0.001, young naive versus CM, and p = 0.06, young naive versus EM). Targets of Sp1 decreased in expression on average (Fig. 3C), but these changes did not achieve statistical significance, possibly because available lists of curated Sp1 targets are less comprehensive than those for the other TFs. Overall, our analyses directly implicate ETS1 and Sp1 as maintaining the naïve state in CD8 T cells, and BATF and Eomes as contributing to the epigenetic changes induced by CD8 T cell differentiation. While this analysis suggests that these TFs are key regulators of the gene expression patterns that define T cell subset identities and functions, other family members sharing their motifs may also serve as important regulators of T cell identity or as redundancy in the transcription network circuitry. The statistical significance of the enrichment of all TF binding motifs is included in Table S1.
Accessibility changes with age in CD8 T cells are linked to TF families involved in memory differentiation
We next investigated whether the TF binding and gene expression changes associated with differentiation were preserved across age cohorts. We found that differentiation in old was similar to differentiation in young, with a strong overlap in the differential peaks and motifs identified (Fig. S3).
To examine age-related dysfunction in the maintenance of T cell states, we compared the chromatin landscapes between young and old individuals within the T cell subsets. We identified 2,864 differentially open peaks (4.5% of all peaks) across these three comparisons (Fig. 4A). Interestingly, the majority of the differentially accessible peaks associated with aging were within the naive and CM subsets, with comparatively few changes observed in the EM.
Fig. 4. Age-associated chromatin openness changes mirror differentiation-associated changes.

(A) Bar graphs of the numbers of differentially open peaks between age cohorts, within each T cell subset. Directions of bars indicate in which age group the peaks were more open.
(B) Boxplot of variation in openness across the six age-segregated T cell subsets, from PCA. The y-axis gives the values for the first PC. Groups are ordered by increasing median.
(C) Boxplot of results from PCA, as in (D), for gene expression from RNA-seq. Cohorts are in the same order as (D), with the original data set in the left panel and an independent validation set in the right panel. For each data set, 0 on the y-axis is set to the median value in the young naive cohort.
(D) k-means clustering of open chromatin peaks that were differentially open between young and old in at least one T cell subset. Peaks are in rows, and subsets are in columns. Colors represent median z-scores of the chromatin openness at a given peak in each subset. Sidebar colors label the three clusters, with the inset numbers giving the numbers of peaks in each cluster. GO analysis of the peaks in the bottom cluster, comprising peaks opening with differentiation, is given on the right. GO enrichment for other clusters is given in Fig. S2B.
(E) k-means clustering, as in (B), of genes differentially expressed between young and old in at least one T cell subset, from RNA-seq data. GO analysis on right is for the dark blue cluster, second from the bottom, which comprised peaks opening with aging.
As verification of the specificity of the peak set we found to be differentially accessible with age, we performed principal component analysis (PCA) on those peaks and evaluated the sample clustering. The samples were effectively separated by age (Figure S4A), but not by donor or sex (Figure S4B), indicating that our results were not meaningfully affected by sample heterogeneity. To assess more directly whether interpersonal variability impacted our estimates of age-associated differences, we quantified their stability by randomly dropping out all possible pairs of samples (one from each age cohort), then rerunning our differential analyses. We found that no single donor drove our results (Figures S4C and S4D). We also compared our findings to those attained by using alternative normalization strategies designed to correct for variability across samples in overall levels of accessibility (36), or in GC and peak-length biases (37). These analyses confirmed that our estimates were highly robust (correlations of fold-change estimates > 0.97, p < 2.2 * 10−16; Figures S4E and S4F).
Using PCA to obtain a broad-based perspective on T cell aging and differentiation, we found that the first PC was sufficient to delimit the six age-specific T cell subsets (Fig. 4B). We observed that the old naive samples were closer to the CM populations than were the young naive, and we noted a similar trend in old CM relative to the EM, indicating that the epigenetic landscape of old T cells is globally biased toward differentiation. Similarly, applying k-means clustering to the peaks changing in accessibility with differentiation, we found that the old cells, and particularly the old naive, attained more effector-like patterns of chromatin structure (Fig. S5A). Furthermore, genes more expressed with age were enriched for GO categories closely aligned with those enriched in genes more expressed following differentiation (p = 4.2 * 10−13, Fisher’s exact test; Fig. S5B).
We found a similar relationship between aging and differentiation in the transcriptome for the naive subset (Fig. 4C). The first PC distinguished the three differentiation stages, and the transcriptome of the old naive was closer to the CM than was the young naive. This differentiation-oriented shift in naive cell gene expression with age was confirmed by RNA-seq of an independent validation set of young and old individuals. In contrast to the ATAC-seq results, no age-associated shift was seen in the CM subset.
When we performed k-means clustering of peaks changing in accessibility with age, we observed that age-associated closures mirrored differentiation-associated closures in the young, with peaks more open in the young naive than young CM and EM uniformly more open in young than old (top two clusters, Fig. 4D). Likewise, peaks more open in EM than naive (bottom cluster, Fig. 4D) increased in openness with age in all three subsets, indicating a widespread progression toward effector-like chromatin accessibility patterns with age. Furthermore, these peaks bordered genes with effector-like GO signatures, associated with pathways related to the regulation of immune response. GO results for the other clusters are shown in Fig. S5C.
The trend of changes in aging resembling those in differentiation was also present in the gene expression levels, but less apparent than in the chromatin accessibility patterns. Applying k-means clustering, we found that genes more expressed in the young naive than young CM and EM declined with age (green cluster, Fig. 4E), and that genes more expressed in EM than naive increased with age (dark blue cluster, Fig. 4E). However, these effects were less striking than in the chromatin accessibility results, and GO analysis of this latter cluster only yielded terms that were of low enrichment and not obviously related to effector function. The observation that the link between aging and differentiation was more evident in chromatin openness than gene expression patterns was unsurprising, as naive and memory T cell populations have been shown to differ significantly in their ability to induce gene expression upon activation, but only minimally in their transcription profiles while in the resting state (12).
To examine whether the drivers of T cell differentiation also contribute to the immune aging of CD8 T cells, we identified the TF binding motifs enriched in loci that changed in accessibility with age. We found high concordance between the enrichment in age-associated and differentiation-associated peaks (Fig. 5A), as was expected because the peak sets overlapped considerably (Fig. S5D). Specifically, the motifs enriched in regions that gained accessibility with age were dominated by bZIPs, notably BATF; in the loci that lost accessibility with age, motifs for ETS and Zf TFs were enriched, with steady-state expression levels of the TFs again nominating ETS1 and Sp1 as having key roles (Fig. 5B).
Figure 5. TF enrichment in peaks of differential openness in aging parallels that in differentiation.

(A) Expression of TFs with binding motifs enriched in loci more accessible in old naïve (Na) cells. The x-axis indicates TF expression fold-change with age, and the y-axis indicates the –log(p-value) of TF binding motif enrichment in loci that gain accessibility with age. Dot size indicates median TF expression in the old Na, and dot color gives the TF family. A list of the statistical significance of the enrichment of all TF binding motifs in differentially open sites is given in Table S1.
(B) As in (A), for TFs with binding motifs enriched in loci less accessible following aging. Here, dot size indicates TF expression level in the young Na.
(C) Expression changes of BATF targets, from published knockdown experiments in human-derived cell lines (25), in young versus old within T cell subsets. p-values give results of Wilcoxon rank-sum tests. Negative values indicate the gene is more expressed in cells from older individuals.
(D) Expression changes of BATF targets, as in (C), for young Na versus old Na, in the validation cohort.
The age-associated enrichment for BATF binding motifs was of particular interest because BATF is an essential regulator of T cell differentiation (25), and its overexpression has been shown to lead to T cell exhaustion (25, 38). We found that BATF target genes were significantly more expressed in old compared to young in every T cell subset (naive, CM, EM: p = 2.48 * 10−7, 6.02 * 10−5, and 0.0028, respectively, Wilcoxon rank-sum tests; Fig. 5C).
Other classical differentiation-associated TFs, including Eomes, TBX21, RUNX1, and PRDM1, appeared to be specific to differentiation and did not exhibit aging-associated binding motif enrichment. Concordant with this finding, we determined that age-driven expression changes were significantly greater for BATF target genes than for Eomes target genes (Fig. S5E). Additionally, canonical effector genes such as IFNG and GZMB did not increase in accessibility with age (Fig. 1B), suggesting that the mechanisms driving aging only partially resemble those underlying antigen-induced differentiation. Overall, our findings indicate that changes in chromatin accessibility patterns, TF binding motif utilization, and gene expression all support the model that naïve and central memory CD8 T cells become more epigenetically primed for memory and effector cell function with age, while losing naive- and central memory-associated features.
Loss of promoter openness during aging associated with decline in NRF1 activity
Regions of greater chromatin accessibility in young tended to be highly open promoter regions, while enhancers constituted a greater percentage of peaks more open in old (Fig. 6A, Fig. S6A). Compared to all regions in our peak set, the ratio of promoters to enhancers was significantly greater for peaks more open in young (p < 10−91, binomial test) and significantly lesser for peaks more open in old (p < 10−60, binomial test; Fig. 6B, upper pane), a disparity which was not attributable to technical biases or systematic imbalances in data quality (Figures S6B–E). None of the comparisons between T cell subsets showed a shift from the overall promoter-to-enhancer ratio as great as that of the aging comparisons (Fig. 6B, lower pane). This discrepancy suggests that loss of chromatin openness at promoters is a unique feature of immune aging.
Fig. 6. Aging is correlated with an erosion of accessibility at promoters.

(A) Violin plots of the distributions of distances of peaks to the nearest transcription start site (from RefSeq annotations). The x-axis scale is in log10 of base pairs.
(B) Fractions of promoters versus enhancers, from ChromHMM annotations in naive CD8 T cells, for sets of peaks with significant openness changes in the age or differentiation comparisons. Fractions are with respect to proportions of peaks. Vertical, red line at 0.53 represents the overall promoter fraction across all peaks.
Because we did not observe a similar shift in the chromatin openness changes associated with differentiation, we hypothesized that a molecular driver of the chromatin signatures of aging would be a TF with a binding motif highly enriched in loci closing with age, but not highly enriched in loci closing with differentiation. We found that NRF1 best matched this pattern (Fig. S7A). Among the top twenty most significantly enriched TF binding motifs from the aging and differentiation comparisons, sites for NRF TFs were the most skewed toward promoter regions relative to enhancers (Fig. 7A; NRF in yellow). Enrichment for NRF1 motifs in loci closing with age was maintained even after controlling for the overall promoter-to-enhancer shift, both for the naive (Fig. S7B) and the CM (Fig. S7C).
Fig. 7. Age-driven loss of NRF1 binding associated with decline in expression of mitochondrial respiration complex (MRC) genes.

(A) Fractions of promoters versus enhancers, from ChromHMM annotations in naive CD8 T cells, for TF families, using peaks with binding motifs for the constituent TFs. The NRF family is highlighted in yellow. Vertical, red line at 0.53 represents the overall promoter fraction across all peaks.
(B) Comparison of aggregate footprints for NRF1 in young versus old naive (Na). Y-axis values are average normalized reads per motif site, per donor, at sites that close with age.
(C) Overlap between NRF1 binding sites from ChIP-seq, ATAC-seq peaks more open in young than old Na, and ATAC-seq peaks not more open in young than old Na. Association between accessibility increases and NRF1 binding: p < 2.2 * 10−16, Fisher’s exact test.
(D) MRC gene expression changes with age in naïve cells. Positive fold-changes indicate the gene is more expressed in the young than in the old. p-value gives result of Wilcoxon rank-sum tests assuming a median log2(fold-change) of 0 (red line).
(E) As in (D), from RNA-seq in an independent validation cohort.
(F) Overlap between NRF1 binding sites from NRF1 ChIP-seq, ATAC-seq peaks, and MRC gene promoters. Association between MRC gene promoters and NRF1 binding: p = 1.5 * 10−12, Fisher’s exact test.
(G) NRF1 footprint, as in (B), for NRF1 knockdown in young naive cells, versus control-transfected naive cells from the same donors. Binding sites used are those from the ChIP-seq results.
(H) Western blot of NRF1 (top row) and β-actin control (bottom row) in NRF1-knockdown naive cells from young donors (right column), versus control-transfected naive cells from the same donors (left column).
(I) As in (D) and (E), for MRC gene expression changes in the NRF1 knockdown versus control. Negative fold-changes indicate the gene decreased in expression following the knockdown.
(J) Oxygen consumption rates (OCR; y-axis) in young Na (light purple) versus old Na (dark purple). Basal OCR (0–20 minutes) and maximal OCR (60–80 minutes, after FCCP treatment) were significantly higher in young than old (p = 0.022 and 0.021, respectively, F-tests). Results are from six young and five older individuals. Points represent mean OCR, with error ranges reflecting standard errors.
(K) Cytometry of MitoTracker Green staining (MFI) in naive cells from four young and four older adults, giving relative mitochondrial mass.
Footprinting the NRF1 motif, we found greater chromatin openness in young naive compared to old naive around NRF1 sites in loci that close with age (Fig. 7B). Using NucleoATAC (39), we determined that aggregate occupancy of nucleosomes at NRF1 sites was 30% greater in the old than in the young (Fig. S7D) – the greatest shift among the TFs with the top 10 most significantly enriched motifs in the naive aging comparison. These results indicated that NRF1 binding activity is a key feature of the epigenetic landscape of young naïve CD8 T cells. To further investigate this inference, we performed NRF1 ChIP-seq in naive CD8 T cells from young donors. Peaks more open in young than old naive were significantly associated with NRF1 binding (p < 2.2 * 10−16, Fisher’s exact test; Fig 7C). This observation confirmed the specificity of NRF1 binding in maintaining chromatin openness at loci characteristic of youth in the naive subset.
NRF1 is a critical regulator of mitochondrial biogenesis (40), specifically mitochondrial respiratory chain complex (MRC) genes (41, 42). MRC promoters closed with age in naïve CD8 T cells at a greater rate than did other loci (p = 6.32 * 10−5, Fisher’s exact test), and MRC genes decreased in expression in naïve cells with age, both in our original cohort (p = 1.3 * 10−4, Wilcoxon rank-sum test; Fig. 7D) and an independent validation set (p = 3.7 * 10−9, Wilcoxon rank-sum test; Fig 7E). Furthermore, NRF1 binding was significantly enriched both at MRC promoters in general (p = 1.5 * 10−12, Fisher’s exact test; Fig 7F), and at those that exhibited a loss in accessibility in the naive cells with age (p = .012, Fisher’s exact test).
NRF1 gene expression decreased slightly, though not significantly, in the naive cells with age (Fig. 5A). To test whether reducing NRF1 expression induces a corresponding loss in NRF1 binding, we knocked down NRF1 in young naive cells. We detected only a minimal decrease in occupancy at NRF1 binding sites (Fig. 7G), despite a substantial decrease in NRF1 protein levels (Fig. 7H). Accordingly, the knockdown induced a decrease in MRC gene expression (p = .002, Wilcoxon rank-sum test; Fig 7I), but one that was more modest than the age-associated difference.
We investigated the functional impact of the drop in MRC gene expression in naive cells with age by comparing the rates of mitochondrial respiration in young and old naïve CD8 T cells. Both basal and maximal oxygen consumption rates (OCR) were decreased in the old naive relative to the young (p = 0.022 and 0.021, respectively, F-tests; Fig. 7J). Interestingly, mitochondrial mass was increased in the old compared to the young (p = 0.029, Wilcoxon rank-sum test; Fig. 7K), resulting in an even larger difference in OCR when normalized to mitochondrial mass. Overall, our findings suggest that the failure of NRF1 to maintain promoter openness with age may compromise the metabolic state of old naive CD8 T cells, resulting in increased cell loss and an inability to mount a vigorous, energy-demanding clonal expansion in immune response.
Discussion
In this study, we generated genome-wide maps of chromatin accessibility in young human CD8 T cell subsets to define the epigenetic landscape of normal CD8 T cell differentiation. We then quantified the changes to this landscape with age. We determined that the naïve state was characterized by increased accessibility at ETS and zinc finger TF binding sites, including those of ETS1 and Sp1, while memory states exhibited greater openness at bZIP and T-box sites, in particular for BATF and Eomes. Chromatin accessibility in effector and central memory was largely conserved. While differentiation-associated chromatin accessibility signatures were mostly preserved during aging, old T cells were epigenetically shifted toward memory differentiation. We further observed an age-associated loss of chromatin accessibility at promoters, accompanying a decrease in occupancy of promoter-binding NRF1 in old naive T cells. Therefore, from an epigenetic perspective, CD8 T cell aging occurs along two dimensions: a shift toward memory-associated chromatin openness patterns and an erosion of accessibility at promoters (Fig. 8).
Fig. 8. Integrated view of changes in chromatin openness and TF binding in aging and differentiation.

General model of chromatin structure and TF binding changes in immune aging and differentiation. The positions of the six age-specific T cell subsets correspond to the groups’ respective centroids in the space of the first two PCs from PCA. Aging occurs along two dimensions: a predisposition toward differentiation (x-axis) and an erosion of accessibility at promoters (y-axis). Differentiation is associated with a drop in accessibility at ETS and Zf binding sites and a commensurate gain at bZIP, T-box, and Runt binding sites. While differentiation is preserved during aging, there is also a loss in binding of promoter-binding TFs such as NRF1, a gain of differentiation-associated bZIP TFs, and a loss of differentiation-associated ETS TFs.
The observed partial differentiation of aged naïve T cells (Figures 4 and S5) is reminiscent of stem cell aging. Stem cells are characterized by their pluripotency and capacity for self-renewal, but in response to DNA damage or proliferative stress, they clonally expand, start to differentiate, and lose their pluripotency. Naïve T cells also need to self-renew via homeostatic proliferation, which leads to telomeric erosion and increasing clonality with age (7). As in stem cells, chronic replicative stress in T cells may be sufficient to activate TF networks associated with differentiation, such as those mediated by BATF. Induction of BATF activity in stem cells, via telomeric stress, has been implicated in age-associated increases in the differentiation and clonality of lymphocyte precursors, and a decline in lymphocyte generation (43). Similarly, in old T cells, decades of homeostatic proliferation may slowly erode the naïve epigenetic signature and push the cells toward a differentiated state. Consequent to this shift, the old T cells might be likewise described as exhibiting a loss in stem-ness, resulting in diminished proliferative potential. Defective homeostatic proliferation would explain the striking loss of naïve CD8 T cells with age. Moreover, a diminished proliferative potential would curtail the rapid expansion of T cells in response to antigenic stimulation. Because our study focused on establishing broad-based TF expression levels and binding motif enrichment, further work specifically focusing on the implicated TFs, such as BATF, will be needed to directly characterize TF binding and functional relevance.
The finding of a more differentiated state in old naive CD8 T cells could be the result of contaminating memory cells. We have used a stringent strategy to purify naïve T cells, but a subset of memory CD8 T cells, termed stem-like memory T cells, reacquire a phenotype close to naïve T cells (44). These T cells can persist for decades after the initial antigen has been cleared and masquerade as naïve cells in the absence of re-exposure. Moreover, naïve CD8 T cells able to produce IFNG and GZMB upon stimulation have been reported to increase in number with age (45). Although some of these cells recognize viral epitopes, they have longer telomeres than do antigen-driven memory cells, suggesting that many have acquired effector function as a consequence of non-antigen-driven proliferation, similar to virtual memory cells in murine systems (4, 46). Several findings argue against the interpretation that our age-associated epigenetic signature reflects contamination by stem-like memory T cells. First, typical memory genes, including IFNG and GZMB, were as inaccessible in old as in young naïve T cells (Fig. 1B). We also explored the possibility of contamination computationally and found that the accessibility levels in the old naive were inconsistent with that population being a mixture of the young naive and young CM subsets (Fig. S8). Finally, a more differentiated chromatin signature was also found for aged central memory T cells (Figures 4B, 4C, and S3A); this cannot be explained by the accumulation of stem-like memory T cells, which could only have contaminated the naïve subset.
The other dimension of immune aging we observed occurred through a loss of chromatin openness at promoters in old T cells. The NRF TF family has previously been shown to be enriched in human promoter sequences (47), and further investigation revealed that the NRF1 motif was strikingly enriched in the promoters that were more open in the young T cells than in the old. Through ChIP-seq of NRF1, we found a significant overlap between NRF1-regulated sequences and sites closing in naïve CD8 T cells with age (Fig 7C). NRF1 binding sites were also enriched at the promoters of MRC genes (Fig 7F), concordant with prior studies demonstrating that NRF1 plays an essential role in regulating mitochondrial gene expression (40, 48). In Seahorse assays, oxygen consumption rates were clearly reduced in naïve CD8 T cells from older individuals (Fig. 7J), despite their having greater mitochondrial mass (Fig. 7K). These findings are consistent with defects in the electron transport chain that could be related to the reduction of MRC gene transcription. However, a decline in NRF1 protein levels was insufficient to explain the reduced chromatin accessibility at NRF1 binding sites and its relationship to the transcription of MRC genes. Further investigation is needed to identify the mechanisms through which NRF1 accessibility is lost with age. Previous work has demonstrated that cofactor-maintained hypomethylation of NRF1 binding sites is essential for the TF to bind (49), suggesting that progressive increases in the methylation of these loci could be a major contributor.
The aging-associated signatures we identified were notably more prominent in our epigenetic data than in our transcriptomic data. This result is concordant with recent findings in mice that activation of naive T cells leads to stable epigenetic priming for memory differentiation (12). The accessibility patterns indicative of activation-induced chromatin remodeling were persistent through replication and were likewise observed in memory populations, though baseline gene expression levels were only minimally affected.
Our results describe CD8 T cell aging as the accumulation of naive cells that are epigenetically primed for memory differentiation. Our study serves as an essential counterpart to previous studies investigating histone modifications associated with aging in humans (11), by elucidating the significant differences in chromatin accessibility and associated TF binding, which reflect shifts in the active regulatory programs. Building on these foundations, future characterization of the epigenetic changes will be required to further our understanding of the functional defects, and the regulatory dysfunction that generates these defects, occurring during immune aging.
Materials and Methods
Study design
This study was designed to characterize the epigenetic landscapes and molecular drivers associated with human CD8 T cell aging, as well as those associated with maintaining the naive and memory states.
Our experimental design utilized repeated measures, with donors as the blocking variable: for every donor, each of the naive, central memory, and effector memory T cell subsets were subjected to ATAC-seq. Having multiple data points for each donor allowed us to statistically control for interpersonal variability in chromatin openness.
Cells were collected from five young (aged 20–35 years) and five elderly adults (aged 60–85 years). All individuals were healthy, regular platelet donors without a history of autoimmune disease, diabetes mellitus, or cancer.
ATAC-seq analysis and peak annotation
We performed ATAC-seq on purified T cell subsets using standard protocols (see Supplemental Methods). We aligned the data to hg19 using Bowtie 2, and called a total of 64,089 peaks with MACS2 (50). Gene and TSS annotations were drawn from RefSeq and GENCODE. ChromHMM calls for naive CD8 T cells were downloaded from the Roadmap Epigenomics Project.
Differential openness and TF binding motif enrichment calls
To assess differential openness, we applied DESeq2, using a model with a main effect for age, an interaction term for age with subset, and an interaction term for age with donors (blocking on donors in each age cohort). Peaks were considered to be differentially open if the corrected p-value was less than 0.1.
For highly significant peaks (corrected p-value < 0.05), we performed k-means clustering using the median z-score for each T cell subset. The number of clusters was determined with the gap statistic. Gene ontology analysis was performed using GREAT (21).
Transcription factor binding motif enrichment was calculated with HOMER (51). TFs of interest were chosen as those that had both enriched motifs and significant changes in expression. TF target sets were drawn from published results on genome-wide expression studies based on knockdown experiments in human-derived cell lines (25, 32–35).
RNA-seq
RNA-seq libraries were constructed according to standard protocols (see Supplemental Methods). The data were aligned with TopHat2 and differential expression was tested using DESeq2. Gene ontology analysis was performed with topGO.
When running PCA, we used the counts from the 1,000 genes with the highest overall variance in the original cohort. The validation cohort was then projected onto these dimensions. To enable direct comparisons between the two cohorts, within each cohort the median PC 1 value of the young naive was subtracted from each sample’s coordinate.
ChIP-seq
ChIP-seq was performed on naïve T cells using two NRF1-specific antibodies, according to standard protocols (see Supplemental Methods). Reads were aligned to the genome using Bowtie 2 and a total of 3,375 peaks were called with MACS2.
Seahorse Assay
The Seahorse assay was performed according to the manufacturer’s instructions (see Supplemental Methods). Statistical significance was assessed by analyzing each time-point as separate repeated measures ANOVA experiments.
Supplementary Material
Acknowledgments
The authors thank B. Wu for technical assistance, A. Schep and S. Denny for data analysis discussion, and R. Ahmed for helpful comments.
Funding
This work was supported by National Institutes of Health (NIH) grants R01 AG015043 and R01 AI108891 (J.J.G.), U19 AI057266 (J.J.G. and W.J.G), P50HG007735 (W.J.G.), and R01 AI108906 (C.M.W), and by the Rita Allen Foundation (W.J.G.), the Baxter Foundation Faculty Scholar Grant (W.J.G.), and a grant from Stanford Bio-X. D.M.M. was supported by the Stanford Biomedical Informatics Training Grant from the National Library of Medicine, LM-07033, and D.W.Z. by the NIH Training Program in Adult and Pediatric Rheumatology Grant, T32AR050942.
Footnotes
This manuscript has been accepted for publication in Science Immunology. This version has not undergone final editing. Please refer to the complete version of record at www.scienceimmunology.org. The manuscript may not be reproduced or used in any manner that does not fall within the fair use provisions of the Copyright Act without the prior, written permission of AAAS.
Epigenetic signatures of CD8 T cell aging include TF networks involved in differentiation and loss of NRF1 site accessibility
Author Contributions
J.J.G., W.J.G., D.M.M., D.W.Z., J.D.B., and C.M.W. designed the research. S.L.S., B.H., R.E.Y., and Z.Y. performed experiments. D.M.M. and D.W.Z. analyzed the data. D.M.M., D.W.Z., W.J.G., and J.J.G. wrote the paper.
Competing interests
W.J.G. is listed as an inventor on a pending patent for ATAC-seq, and is a cofounder and scientific advisor of Epinomics.
Data deposition and materials availability
Sequencing data are available in dbGaP, accession phs001187.v1.p1. Raw data for Figures 7J and 7K are available in Table S2.
Supplementary Materials
Supplementary Methods
Figure S1: Isolation of CD8 T cell subsets by cell sorting.
Figure S2: Relationship of differentially open peaks in CD8 T cell differentiation with gene pathways and expression.
Figure S3: Correlation between differentiation-associated chromatin openness changes in young individuals and changes in old individuals.
Figure S4: Robustness analyses of estimates of chromatin accessibility changes with age.
Figure S5: Chromatin openness and gene expression patterns associated with aging resemble those of differentiation.
Figure S6: Aging is associated with an erosion of accessibility at promoters.
Figure S7: NRF1 binding activity maintains the young CD8 T cell epigenetic landscape.
Figure S8: Distribution of per-peak estimates of proportions of young CM cells in old naive.
Table S1: TF binding motif enrichment.
Table S2: Data values for oxygen consumption rates and mitochondrial mass.
References and Notes
- 1.Thompson WW. Mortality Associated With Influenza and Respiratory Syncytial Virus in the United States. JAMA. 2003;289:179. doi: 10.1001/jama.289.2.179. [DOI] [PubMed] [Google Scholar]
- 2.Levin MJ. Immune senescence and vaccines to prevent herpes zoster in older persons. Current Opinion in Immunology. 2012;24:494–500. doi: 10.1016/j.coi.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 3.Goronzy JJ, Weyand CM. Understanding immunosenescence to improve responses to vaccines. Nature Immunology. 2013;14:428–436. doi: 10.1038/ni.2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nikolich-Žugich J. Aging of the T cell compartment in mice and humans: from no naive expectations to foggy memories. J Immunol. 2014;193:2622–2629. doi: 10.4049/jimmunol.1401174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.den Braber I, et al. Maintenance of peripheral naive T cells is sustained by thymus output in mice but not humans. Immunity. 2012;36:288–297. doi: 10.1016/j.immuni.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 6.Westera L, et al. Lymphocyte maintenance during healthy aging requires no substantial alterations in cellular turnover. Aging Cell. 2015;14:219–227. doi: 10.1111/acel.12311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qi Q, et al. Diversity and clonal selection in the human T-cell repertoire. Proceedings of the National Academy of Sciences. 2014;111:13139–13144. doi: 10.1073/pnas.1409155111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wertheimer AM, et al. Aging and Cytomegalovirus Infection Differentially and Jointly Affect Distinct Circulating T Cell Subsets in Humans. The Journal of Immunology. 2014;192:2143–2155. doi: 10.4049/jimmunol.1301721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goronzy JJ, Fang F, Cavanagh MM, Qi Q, Weyand CM. Naive T Cell Maintenance and Function in Human Aging. The Journal of Immunology. 2015;194:4073–4080. doi: 10.4049/jimmunol.1500046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Han S, Brunet A. Histone methylation makes its mark on longevity. Trends in Cell Biology. 2012;22:42–49. doi: 10.1016/j.tcb.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The Hallmarks of Aging. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bevington SL, et al. Inducible chromatin priming is associated with the establishment of immunological memory in T cells. Embo j. 2016;35:515–535. doi: 10.15252/embj.201592534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Araki Y, et al. Genome-wide Analysis of Histone Methylation Reveals Chromatin State-Based Regulation of Gene Transcription and Function of Memory CD8+ T Cells. Immunity. 2009;30:912–925. doi: 10.1016/j.immuni.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scharer CD, Barwick BG, Youngblood BA, Ahmed R, Boss JM. Global DNA Methylation Remodeling Accompanies CD8 T Cell Effector Function. The Journal of Immunology. 2013;191:3419–3429. doi: 10.4049/jimmunol.1301395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Russ BE, et al. Distinct Epigenetic Signatures Delineate Transcriptional Programs during Virus-Specific CD8+ T Cell Differentiation. Immunity. 2014;41:853–865. doi: 10.1016/j.immuni.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buenrostro JD, Wu B, Chang HY, Greenleaf WJ. Current Protocols in Molecular Biology. Wiley-Blackwell; 2015. pp. 21.29.21–21.29.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ernst J, Kellis M. ChromHMM: automating chromatin-state discovery and characterization. Nature Methods. 2012;9:215–216. doi: 10.1038/nmeth.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qu K, et al. Individuality and variation of personal regulomes in primary human T cells. Cell Syst. 2015;1:51–61. doi: 10.1016/j.cels.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15 doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McLean CY, et al. GREAT improves functional interpretation of cis-regulatory regions. Nature Biotechnology. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zaret KS, Carroll JS. Pioneer transcription factors: establishing competence for gene expression. Genes & Development. 2011;25:2227–2241. doi: 10.1101/gad.176826.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nature Reviews Immunology. 2012;12:749–761. doi: 10.1038/nri3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Intlekofer AM, et al. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nature Immunology. 2005;6:1236–1244. doi: 10.1038/ni1268. [DOI] [PubMed] [Google Scholar]
- 25.Kurachi M, et al. The transcription factor BATF operates as an essential differentiation checkpoint in early effector CD8+ T cells. Nature Immunology. 2014;15:373–383. doi: 10.1038/ni.2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsukumo Si, et al. Bach2 maintains T cells in a naive state by suppressing effector memory-related genes. Proceedings of the National Academy of Sciences. 2013;110:10735–10740. doi: 10.1073/pnas.1306691110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rutishauser RL, et al. Transcriptional repressor Blimp-1 promotes CD8(+) T cell terminal differentiation and represses the acquisition of central memory T cell properties. Immunity. 2009;31:296–308. doi: 10.1016/j.immuni.2009.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crotty S, Johnston RJ, Schoenberger SP. Effectors and memories: Bcl-6 and Blimp-1 in T and B lymphocyte differentiation. Nat Immunol. 2010;11:114–120. doi: 10.1038/ni.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McLane LM, et al. Differential localization of T-bet and Eomes in CD8 T cell memory populations. J Immunol. 2013;190:3207–3215. doi: 10.4049/jimmunol.1201556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muthusamy N, Barton K, Leiden JM. Defective activation and survival of T cells lacking the Ets-1 transcription factor. Nature. 1995;377:639–642. doi: 10.1038/377639a0. [DOI] [PubMed] [Google Scholar]
- 31.Anderson MK, Hernandez-Hoyos G, Diamond RA, Rothenberg EV. Precise developmental regulation of Ets family transcription factors during specification and commitment to the T cell lineage. Development. 1999;126:3131–3148. doi: 10.1242/dev.126.14.3131. [DOI] [PubMed] [Google Scholar]
- 32.Teo AKK, et al. Pluripotency factors regulate definitive endoderm specification through eomesodermin. Genes & Development. 2011;25:238–250. doi: 10.1101/gad.607311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Verschoor ML, Verschoor CP, Singh G. Ets-1 global gene expression profile reveals associations with metabolism and oxidative stress in ovarian and breast cancers. Cancer Metab. 2013;1:17. doi: 10.1186/2049-3002-1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mansilla S, Priebe W, Portugal J. Changes in gene expression induced by Sp1 knockdown differ from those caused by challenging Sp1 binding to gene promoters. Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms. 2011;1809:327–336. doi: 10.1016/j.bbagrm.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 35.Nikulenkov F, et al. Insights into p53 transcriptional function via genome-wide chromatin occupancy and gene expression analysis. Cell Death and Differentiation. 2012;19:1992–2002. doi: 10.1038/cdd.2012.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Denny SK, et al. Nfib Promotes Metastasis through a Widespread Increase in Chromatin Accessibility. Cell. 2016;166:328–342. doi: 10.1016/j.cell.2016.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hansen KD, Irizarry RA, Wu Z. Removing technical variability in RNA-seq data using conditional quantile normalization. Biostatistics. 2012;13:204–216. doi: 10.1093/biostatistics/kxr054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Quigley M, et al. Transcriptional analysis of HIV-specific CD8+ T cells shows that PD-1 inhibits T cell function by upregulating BATF. Nature Medicine. 2010;16:1147–1151. doi: 10.1038/nm.2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schep AN, et al. Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions. Genome Research. 2015;25:1757–1770. doi: 10.1101/gr.192294.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gleyzer N, Vercauteren K, Scarpulla RC. Control of Mitochondrial Transcription Specificity Factors (TFB1M and TFB2M) by Nuclear Respiratory Factors (NRF-1 and NRF-2) and PGC-1 Family Coactivators. Molecular and Cellular Biology. 2005;25:1354–1366. doi: 10.1128/MCB.25.4.1354-1366.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Finck BN, et al. Lipin 1 is an inducible amplifier of the hepatic PGC-1α/PPARα regulatory pathway. Cell Metabolism. 2006;4:199–210. doi: 10.1016/j.cmet.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 42.Scarpulla RC. Nuclear Control of Respiratory Chain Expression by Nuclear Respiratory Factors and PGC-1 -Related Coactivator. Annals of the New York Academy of Sciences. 2008;1147:321–334. doi: 10.1196/annals.1427.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang J, et al. A Differentiation Checkpoint Limits Hematopoietic Stem Cell Self-Renewal in Response to DNA Damage. Cell. 2012;148:1001–1014. doi: 10.1016/j.cell.2012.01.040. [DOI] [PubMed] [Google Scholar]
- 44.Fuertes Marraco SA, et al. Long-lasting stem cell-like memory CD8+ T cells with a naive-like profile upon yellow fever vaccination. Science Translational Medicine. 2015;7:282ra248–282ra248. doi: 10.1126/scitranslmed.aaa3700. [DOI] [PubMed] [Google Scholar]
- 45.Pulko V, et al. Human memory T cells with a naive phenotype accumulate with aging and respond to persistent viruses. Nat Immunol. 2016;17:966–975. doi: 10.1038/ni.3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Akue AD, Lee JY, Jameson SC. Derivation and maintenance of virtual memory CD8 T cells. J Immunol. 2012;188:2516–2523. doi: 10.4049/jimmunol.1102213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.FitzGerald PC. Clustering of DNA Sequences in Human Promoters. Genome Research. 2004;14:1562–1574. doi: 10.1101/gr.1953904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu Z, et al. Mechanisms Controlling Mitochondrial Biogenesis and Respiration through the Thermogenic Coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 49.Domcke S, et al. Competition between DNA methylation and transcription factors determines binding of NRF1. Nature. 2015;528:575–579. doi: 10.1038/nature16462. [DOI] [PubMed] [Google Scholar]
- 50.Zhang Y, et al. Model-based Analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Heinz S, et al. Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Molecular Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gray KA, Yates B, Seal RL, Wright MW, Bruford EA. Genenames.org: the HGNC resources in 2015. Nucleic Acids Res. 2015;43:D1079–D1085. doi: 10.1093/nar/gku1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.