

Significance
Chronic mucocutaneous candidiasis (CMC) is defined as persistent or recurrent infections of the skin and/or mucosae by commensal fungi of the Candida genus. It is often seen in patients with T-cell deficiencies, whether inherited or acquired, who typically suffer from multiple infectious diseases. Rare patients are otherwise healthy and display isolated CMC, which often segregates as a Mendelian trait. In 2011, we described the first genetic cause of isolated CMC, with autosomal recessive (AR), complete IL-17 receptor A (IL-17RA) deficiency, in a single patient. We report here 21 patients from 12 unrelated kindreds, homozygous for 12 different mutant alleles that underlie AR IL-17RA deficiency. All patients have isolated CMC and their cells do not respond to IL-17A, -17F, and -17E/IL-25.
Keywords: genetics, immunodeficiency, candidiasis
Abstract
Chronic mucocutaneous candidiasis (CMC) is defined as recurrent or persistent infection of the skin, nails, and/or mucosae with commensal Candida species. The first genetic etiology of isolated CMC—autosomal recessive (AR) IL-17 receptor A (IL-17RA) deficiency—was reported in 2011, in a single patient. We report here 21 patients with complete AR IL-17RA deficiency, including this first patient. Each patient is homozygous for 1 of 12 different IL-17RA alleles, 8 of which create a premature stop codon upstream from the transmembrane domain and have been predicted and/or shown to prevent expression of the receptor on the surface of circulating leukocytes and dermal fibroblasts. Three other mutant alleles create a premature stop codon downstream from the transmembrane domain, one of which encodes a surface-expressed receptor. Finally, the only known missense allele (p.D387N) also encodes a surface-expressed receptor. All of the alleles tested abolish cellular responses to IL-17A and -17F homodimers and heterodimers in fibroblasts and to IL-17E/IL-25 in leukocytes. The patients are currently aged from 2 to 35 y and originate from 12 unrelated kindreds. All had their first CMC episode by 6 mo of age. Fourteen patients presented various forms of staphylococcal skin disease. Eight were also prone to various bacterial infections of the respiratory tract. Human IL-17RA is, thus, essential for mucocutaneous immunity to Candida and Staphylococcus, but otherwise largely redundant. A diagnosis of AR IL-17RA deficiency should be considered in children or adults with CMC, cutaneous staphylococcal disease, or both, even if IL-17RA is detected on the cell surface.
Chronic mucocutaneous candidiasis (CMC) is characterized by chronic infections of the skin, nails, and oropharyngeal and genital mucosae caused by Candida albicans. It affects patients with various acquired T-cell immunodeficiencies, including HIV infection, who typically suffer from multiple infections. Inherited forms of CMC are less common and are often associated with other infectious and noninfectious complications, particularly in patients with profound T-cell deficits (1). Patients with autosomal dominant (AD) hyper-IgE syndrome (HIES), caused by heterozygous dominant negative mutations of STAT3, display fewer infections, and patients with autosomal recessive (AR) autoimmune polyendocrine syndrome type 1 (APS-1) are not prone to other infections (2, 3). Finally, rare patients with inherited but idiopathic forms of CMC, referred to as CMC disease (CMCD), have been described since the late 1960s (4–8). These patients may display isolated CMC, but they often also display cutaneous staphylococcal disease (nonetheless referred to as CMCD) or other infectious and/or autoimmune clinical manifestations (syndromic CMCD).
The genetic causes of CMCD described to date include AR IL-17RA deficiency in a single patient (9), AD IL-17F deficiency in a multiplex kindred (9), AR IL-17RC deficiency in three kindreds (10), and AR ACT1 deficiency in a multiplex kindred (ACT1 is a cytosolic adapter of IL-17 receptors) (11). IL-17RA and -17RC belong to the IL-17 receptor family, which also includes the IL-17RB, -17RD, and -17RE chains. These receptors form various heterodimers, through which different IL-17 cytokines signal in an ACT1-dependent manner (12). Finally, AD signal transducer and activator of transcription 1 (STAT1) gain of function (GOF) was reported in ∼350 patients with syndromic CMCD (13–51) and found in approximately half of such patients in our study cohort. In patients with STAT1 GOF mutations, CMC results, at least partly, from impairment of the development and/or survival of IL-17A/F–producing T cells, the underlying mechanisms of which remain unknown (28, 52). Patients with these mutations, who had long been known to be prone to thyroid autoimmunity, were recently found to display other infectious and autoimmune phenotypes (16, 17, 23, 37, 51). Another genetic etiology of syndromic CMCD has recently been described, with AR retinoic acid-related orphan receptors γ (ROR-γ/γT) deficiency in three kindreds with CMC and severe mycobacterial disease (53).
AD HIES and AR APS-1 can, thus, also be seen as syndromic forms of CMCD. Alternatively, STAT1 GOF and ROR-γ/γT deficiency can be seen as distinct entities, separate from CMCD. In either case, impaired IL-17A/F– or IL-17RA/RC–dependent immunity is the core mechanism accounting for CMC in patients with any of these eight inherited disorders. Indeed, all patients with inborn errors of IL-17F, -17RA, -17RC, or ACT1 display CMC. These patients display dysfunctional IL-17F and -17A/F (IL-17F mutations) or dysfunctional responses to IL-17A, -17A/F, and -17F (mutations in IL-17RA, -17RC, and ACT1). In patients with AD HIES (54–57), AD STAT1 GOF (13, 18, 21, 27–29, 32, 35, 36, 38, 39, 41, 42, 45, 46, 49), or AR ROR-γ/γT deficiency (53), the development of IL-17A/F–producing T cells is impaired. Finally, patients with AR APS-1 have high titers of neutralizing auto-Abs against IL-17A and -17F (58, 59).
The pathogenesis of staphylococcal disease in CMCD patients is less clear. Staphylococcal skin disease is frequently observed in patients with ACT1 and IL-17RA deficiencies, but has not been reported in patients with IL-17F and -17RC deficiencies (9–11, 60). This observation suggests that staphylococcal disease may be partly due to an impairment of IL-17E/IL-25 responses, which normally require IL-17RA and ACT1, but neither IL-17F nor IL-17RC. However, too few patients have been described to draw firm conclusions. In particular, AR IL-17RA deficiency has been described in a single patient with CMC and cutaneous staphylococcal disease (9). We used a genome-wide approach based on whole-exome sequencing (WES) to identify 20 new patients, from 11 unrelated kindreds, bearing homozygous IL17RA mutations. Functional characterization of these variants showed them to be responsible for complete AR IL-17RA deficiency. We also characterized the associated clinical phenotype of the 21 patients, including the patient reported in 2011, encompassing not only CMC and staphylococcal skin infections, but also bacterial infections of the respiratory tract.
Results
Clinical Reports.
We investigated 21 patients with early onset, unexplained CMC (Fig. 1A). The patients originated from Morocco (kindred A), Turkey (kindreds B, C, D, E, K, and L), Japan (kindred F), Saudi Arabia (kindreds G and J), Algeria (kindred H), and Argentina (kindred I). The clinical features of patient 1 (P1) (kindred A), born to first cousins from Morocco, have already been reported (9). The 12 families were unrelated, and 11 were known to be consanguineous. All patients displayed CMC before the age of 6 mo, and 14 patients had also suffered from recurrent staphylococcal skin infections by the same age. CMC affected the skin (intertrigo), the scalp, mucosal sites (oral thrush; anogenital candidiasis), or nails (Table 1). These episodes were effectively managed or prevented with a combination of oral (fluconazole) and topical (nystatin) antifungal treatments. Staphylococcal skin infections were reported in 14 patients suffering from abscesses, folliculitis, furunculosis, and crusted pustules on the face and scalp, sometimes spreading to the shoulders and arms. In addition to these skin infections, eight children also had other recurrent infections, including otitis, sinusitis, bronchitis, and lobar pneumonia. Infections typically responded to antibiotics, but subsequently recurred. P2 and P4 were also treated for suspected pulmonary tuberculosis and tuberculous meningitis, respectively, without microbiological confirmation. None of the other clinical manifestations previously reported in patients with GOF STAT1 mutations, such as autoimmune endocrinopathy, aneurysms, or mucosal carcinomas, were detected (16, 17, 23, 37, 51). Detailed phenotyping of lymphocyte subsets was performed for patients from kindreds D, E, and H and revealed no abnormality (Fig. S1).
Fig. 1.

The 12 kindreds with AR IL-17RA deficiency. (A) Pedigree of 12 families, with their genotypes. Kindred A has already been reported elsewhere (9). E? indicates individuals whose genetic status could not be evaluated; m, mutation. (B) Schematic diagram of the IL-17RA protein, with its extracellular (EC), transmembrane (TM), intracellular (IC), and SEFIR [SEF (similar expression to fibroblast growth factor genes) and IL-17R] domains and the positions affected by the mutations.
Table 1.
Clinical characteristics of the 21 patients with AR IL-17RA deficiency
| Patient (kindred) | Age at diagnosis | Genotype [CADD score] | Sex | Consan-guinity | Origin | Mucocutaneous features | Other clinical features |
| P1 (9) (A) | 1 mo | c.850C > T p.Q284X [40] | M | Yes | Morocco (living in France) | Skin, nails and oral mucosal candidiasis Skin pustules, folliculitis | No |
| P2 (B) | 18 mo | c.256C > T p.Q86X [30] | F | Yes | Turkey | Genital and oral mucosal candidiasis | Suspected pulmonary tuberculosis |
| P3 (C) | 1 mo | c.1302_1318dup p.N440Rfs*50 [22] | F | Yes | Turkey (living in France) | Scalp, genital and oral mucosal candidiasis Skin pustules, folliculitis | Eczema |
| P4 (C) | 2 mo | c.1302_1318dup p.N440Rfs*50 [22] | M | Yes | Turkey (living in France) | Genital and oral mucosal candidiasis Skin pustules, folliculitis | Eczema, suspected tuberculous meningitis, lobar pneumonia |
| P5 (D) | 9 y | c.1159G > A p.D387N [33] | M | Yes | Turkey | Scalp and oral mucosal candidiasis Skin pustules, folliculitis, furunculosis, seborrheic dermatitis | Sinusitis, lobar pneumonia |
| P6 (D) | 4 y | c.1159G > A p.D387N [33] | F | Yes | Turkey | Scalp, genital and oral mucosal candidiasis Skin pustules, furunculosis, seborrheic dermatitis | Conjunctivitis |
| P7 (E) | 1.5 y | c.166_169dup p.C57Yfs*5 [34] | F | Yes | Turkey | Skin, scalp, nails, genital and oral mucosal candidiasis Skin pustules, folliculitis, furunculosis | Sinusitis |
| P8 (E) | 1 y | c.166_169dup p.C57Yfs*5 [34] | M | Yes | Turkey | Skin, scalp, nails and oral mucosal candidiasis Skin pustules, folliculitis, furunculosis | Sinusitis, conjunctivitis |
| P9 (F) | 8 y | c.196C > T p.R66X [14] | F | No | Japan | Oral mucosal candidiasis Folliculitis | Eczema, bronchitis, lobar pneumonia |
| P10 (F) | 6 y | c.196C > T p.R66X [14] | M | No | Japan | Skin, scalp and oral mucosal candidiasis Folliculitis | Eczema, bronchitis, lobar pneumonia |
| P11 (G) | 25 y | c.112_119del p.H38Afs*15 [34] | M | Yes | Saudi Arabia | Oral mucosal candidiasis | No |
| P12 (G) | 15 y | c.112_119del p.H38Afs*15 [34] | F | Yes | Saudi Arabia | Oral mucosal candidiasis | No |
| P13 (H) | 1 mo | c.163+1G > A [25] | F | Yes | Algeria | Skin and genital mucosal candidiasis | No |
| P14 (I) | 1 y | c.1152C > A p.Y384X [38] | M | Yes | Argentina | Skin and oral mucosal candidiasis Skin pustules, folliculitis, furunculosis, abscess | Sinusitis, otitis, lobar pneumonia |
| P15 (J) | 4 y | c.268del p.L90Cfs*30 [23] | F | Yes | Saudi Arabia | Skin, genital and oral mucosal candidiasis | No |
| P16 (J) | 2 y | c.268del p.L90Cfs*30 [23] | M | Yes | Saudi Arabia | Skin, scalp, nails, genital and oral mucosal candidiasis | No |
| P17 (J) | 15 y | c.268del p.L90Cfs*30 [23] | M | Yes | Saudi Arabia | Skin, scalp and oral mucosal candidiasis Folliculitis, furunculosis | No |
| P18 (J) | 10 y | c.268del p.L90Cfs*30 [23] | M | Yes | Saudi Arabia | Skin, scalp and oral mucosal candidiasis Folliculitis, furunculosis | No |
| P19 (K) | 22 y | c.1770_1771dup p.Y591Sfs*29 [26.7] | M | Yes | Turkey | Skin, scalp, nails and oral mucosal candidiasis Skin pustules | Otitis |
| P20 (L) | 13 y | c.769_773del p.P257Rfs*16 [28] | F | Yes | Turkey | Oral mucosal candidiasis Skin abscess | No |
| P21 (L) | 11 y | c.769_773del p.P257Rfs*16 [28] | F | Yes | Turkey | Oral mucosal candidiasis | No |
Fig. S1.

Immunophenotyping. (A) Frequency of naïve (CD45RA+CCR7+), central memory (CD45RA−CCR7+), effector memory (CD45RA−CCR7−), and EMRA (CD45RA+CCR7−) cells among the CD4+ and CD8+ T cells of patients (n = 5), healthy relatives (n = 6), and controls (n = 28). (B) Frequency of CD27+ memory cells in the B-cell compartment of patients (n = 5), healthy relatives (n = 6) and controls (n = 23). (C) Frequency of Treg (FoxP3+CD25+) among CD4+ T cells and frequency of γδ T cells (CD3+TCRγδ +) and mucosal-associated invariant T (CD161+Va7.2+) cells among the total CD3+ T cells of patients (n = 5), healthy relatives (n = 6), and controls (n = 23). (D) Frequency of T helper (Th) subsets within the CD4+ memory compartment of patients (n = 3), healthy relatives (n = 6), and controls (n = 28). Subsets were defined as follows: Tfh (CXCR5+), Th1 (CXCR5−CXCR3+CCR4−CCR6−), Th2 (CXCR5−CXCR3−CCR4+CCR6−), Th1* (CXCR5−CXCR3+CCR4−CCR6+), and Th17 (CXCR5−CXCR3−CCR4+CCR6+). (E) Immunophenotyping of NK cells showing total NK cell frequency in lymphocytes (Left), the frequency of CD56bright cells within the NK cell compartment (Center), and the terminal differentiation profile of the CD56dim compartment (Right), as assessed by the distribution of NKG2A and KIR on cells from patients (n = 5), healthy relatives (n = 6–8), and controls (n = 23). Horizontal bars represent the median value. Ctl, controls; Pat, patients; Rel, relatives.
Mutations in IL17RA.
WES was performed for all patients and led to the detection of biallelic IL17RA mutations, which were confirmed by Sanger sequencing (Fig. 1A). No nonsynonymous coding sequence mutations were identified in the other five genes implicated in CMCD (IL17F, IL17RC, ACT1, STAT1, and RORC) or in any of the genes known to underlie related primary immunodeficiencies, including APS-1 and AD HIES. As shown in Fig. 1B, the only essential splice variant, three nonsense and four frameshift variants (and the corresponding premature stop codons), were located upstream from the segment encoding the transmembrane domain of IL-17RA. By contrast, the p.Y384X nonsense, p.D387N missense, and p.N440Rfs*50 frameshift variants affected the intracellular SEFIR (SEF/IL-17R) domain of IL-17RA, which is required for ACT1 recruitment, whereas the p.Y591Sfs*29 frameshift variant was located in the SEFEX domain (SEFIR extension domain) (61, 62). The healthy parents and siblings tested were all heterozygous for the mutant alleles or homozygous for the wild-type allele, consistent with an AR mode of inheritance with full clinical penetrance. None of the 12 mutant alleles were found in any of the various public databases (Exome Aggregation Consortium, Human Gene Mutation Database, Ensembl, National Heart, Lung, and Blood Institute Grand Opportunity Exome Sequencing Project, and 1000 Genomes Project), our in-house WES database (>3,000 exomes), or the Greater Middle Eastern Variome (63), further suggesting that the mutant alleles were causal for CMCD. The p.D387N missense mutation affected a residue conserved throughout evolution. As expected, combined annotation dependent depletion (CADD) scores predicted all mutations to be deleterious and were well above the mutation significance cutoff score for IL17RA (Table 1) (64, 65). These data strongly suggested that the 21 patients suffered from AR IL-17RA deficiency.
Expression and Function of the Mutant IL17RA Alleles.
IL-17RA expression was tested on the surface of primary or SV40-transformed fibroblasts and/or lymphocyte subsets and monocytes from seven patients homozygous for six mutant IL17RA alleles (Fig. 2). IL-17RA expression was abolished on fibroblasts (P1, P2, and P4) (Fig. 2A) and peripheral blood mononuclear cells (PBMCs) [P1 (9), P3, and P13] (Fig. 2C), except for those from P5 (p.D387N), for whom IL-17RA was barely and normally detectable in SV40 fibroblasts and monocytes, respectively (Fig. 2 A and C). In addition, the p.Y591Sfs*29 allele (P19) was normally expressed in primary fibroblasts (Fig. 2B). The intracellular D387 residue is highly conserved and located in the SEFIR domain, which engages in homotypic dimerization with the SEFIR domain of ACT1 for IL-17RA signaling. We therefore tested HEK293T cells overproducing the p.D387N protein for interactions of this protein with ACT1, by immunoprecipitation and Western blotting; we found that the interaction of these two proteins was severely impaired in these cells (Fig. S2). We then investigated the function of p.D387N, together with several loss-of-expression alleles, by stimulating patient fibroblasts with high doses of recombinant IL-17A, -17F, and -17A/IL-17F heterodimers, with or without the addition of TNF-α. We detected no induction of IL-6 and GRO-α in any condition, whereas the induction of these two proteins was observed in control cells (Fig. 3 and Fig. S3 A and B). We measured the induction of mRNA for the antimicrobial peptide BD2 (β-defensin 2) in patient fibroblasts stimulated with IL-17A plus TNF-α. We found no up-regulation in cells homozygous for p.D387N or p.Q284X, whereas such induction was observed in control cells (Fig. S3C). We then tested the response to IL-17E/IL-25 in the presence of IL-2 in PBMCs from P5 and P6 (p.D387N). No induction of IL-5 was observed, in contrast to the results obtained for control PBMCs (Fig. S4). Finally, the transfection of fibroblasts from P1 and P5 with a WT IL-17RA–encoding vector partially restored both surface IL-17RA expression (Fig. S5) and the response to IL-17A plus TNF-α (Fig. 4 and Fig. S6). Overall, these data indicate that p.D387N is loss of function. All patients displayed complete AR IL-17RA deficiency, with a lack of cellular responses to IL-17A, -17F, and -17A/F in fibroblasts, as well as to IL-17E/IL-25 in PBMCs.
Fig. 2.

IL-17RA expression. (A and B) IL-17RA expression in SV40-immortalized (A) and primary (B) fibroblasts from healthy controls and patients. (C) IL-17RA expression in T cells (CD3+), B cells (CD19+), natural killer cells (CD3−CD56+), and monocytes (CD14+) from healthy controls and three patients. Isotype control, dashed lines; IL-17RA–specific antibody, solid lines.
Fig. S2.

The p.D387N IL-17RA allele impairs IL-17RA/ACT1 interaction. HEK293 cells were transiently transfected with a plasmid encoding FLAG-tagged ACT1 together with plasmids encoding V5-tagged WT–IL-17RA or D387N–IL-17RA. Whole-cell lysates (WCL) were subjected to immunoprecipitation with anti-FLAG antibody. Western blot analysis was performed with anti-FLAG and -V5 antibodies.
Fig. 3.

Function of the mutant IL-17RA alleles. IL-6 and GRO-α fold induction measured in the supernatants of SV40-immortalized fibroblasts from two healthy controls and five patients, after 24 h of stimulation with IL-17A, -17F, -17A/F (100 ng/mL), or TNF-α (20 ng/mL), as assessed by ELISA. Means of three independent experiments are shown. Error bars represent the SD.
Fig. S3.

Function of the mutant IL-17RA alleles. IL-6 and GRO-α production, measured by ELISA in the supernatants of SV40-immortalized (A) and primary fibroblasts (B) from two healthy controls and patients, after 24 h of stimulation with IL-17A, -17F, -17A/F (100 ng/mL), and TNF-α (20 ng/mL) alone or in combination. Means of three technical replicates are shown. Error bars represent the SD. (C) BD2 mRNA induction in SV40-immortalized fibroblasts from two healthy controls and from P1 and P5 after 24 h of stimulation with IL-17A (100 ng/mL) alone or with TNF-α (20 ng/mL). Means of three technical replicates are shown. Error bars represent the SD. Data are representative of two independent experiments. NS, not stimulated.
Fig. S4.

The p.D387N allele is loss of function for the response to IL-17E/IL-25. PBMCs from three controls, P5, P6, and two healthy relatives were cultured with thymic stromal lymphopoietin for 24 h, harvested, and stimulated with IL-2 and -17E/IL-25 for an additional 72 h. IL-5 was determined in the culture supernatants by ELISA. Error bars represent the SD. NS, not stimulated.
Fig. S5.

Complementation of IL-17RA deficiency. IL-17RA expression in SV40-immortalized fibroblasts from a control, P1, and P5 transfected with the empty pORF9mcs plasmid or the pORF9-hIL17RA plasmid. Isotype control, dashed lines; anti–IL-17RA antibody, solid lines.
Fig. 4.

Complementation of IL-17RA deficiency. IL-6 production, measured by ELISA, in the supernatants of SV40-immortalized fibroblasts from a control, P1, and P5, after transfection with the empty pORF9mcs plasmid or the pORF9-hIL17RA plasmid, after 24 h of stimulation with IL-17A (100 ng/mL) alone or in combination with TNF-α (20 ng/mL) is shown. Means of three technical replicates are shown. Error bars represent the SD. One experiment representative of the three carried out is shown. NS, not stimulated.
Fig. S6.
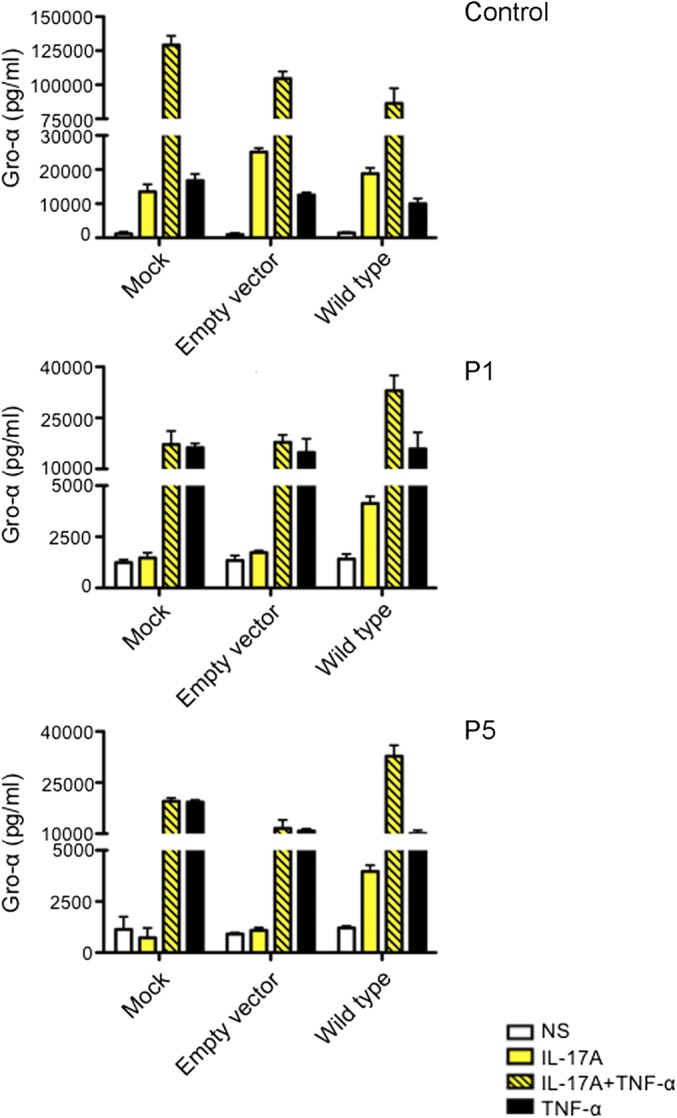
GRO-α production, measured by ELISA, in the supernatants of SV40-immortalized fibroblasts from a control, P1, and P5 after transfection with the empty pORF9mcs plasmid or the pORF9-hIL17RA plasmid and stimulation for 24 h with IL-17A (100 ng/mL) alone or with TNF-α (20 ng/mL). Means of three technical replicates are shown. Error bars represent the SD. One experiment representative of the three carried out is shown. NS, not stimulated.
Abnormally High Proportions of IL-17–Producing T Cells and a Normal Response of Whole Blood to Candida and Staphylococcus.
Given the critical role of IL-17A/F–producing T cells in immunity to Candida at barrier sites, we carried out an ex vivo assessment of the proportions of IL-17A/F–producing memory CD4+ T cells in patients. The patients tested (kindreds C, D, E, and H) had significantly higher proportions of IL-17A– and IL-17F–producing memory CD4+ T cells ex vivo than controls and healthy relatives, after stimulation with phorbol 12-myristate 13-acetate (PMA) and ionomycin, but similar or slightly higher proportions of IL-22–producing memory CD4+ T cells (Fig. 5A). By contrast, the mean values for IL-17A and -22 secretion levels in whole-blood assays were slightly higher than those for controls and healthy relatives, although this difference was not significant. This difference probably resulted from the smaller numbers of memory CD4+ T cells in patients than in adult controls and healthy relatives (Fig. 5B and Fig. S1A). We also carried out whole-blood assays to assess the response to different stimuli, including zymosan, curdlan, lipopolysaccharide (LPS), vesicular stomatitis virus (VSV), Bacille de Calmette et Guerin (bacillus Calmette–Guérin), Staphylococcus aureus, and yeasts (C. albicans, Saccharomyces cerevisiae, and Exophiala dermatitidis), by measuring the secretion of IL-6, -17A, and IFN-γ. Similar responses were observed for controls, the patients, and their healthy heterozygous relatives (Fig. S7). The results of these two sets of experiments suggest that the reported infectious phenotype in patients cannot be assigned to a defect in the mounting of a potent IL-17 inflammatory response or in the response to S. aureus and C. albicans. Instead, they suggest that the susceptibility to S. aureus and C. albicans reported in IL-17RA–deficient patients results from a complete lack of response to at least IL-17A, -17F, -17A/F, and -17E/IL-25.
Fig. 5.

IL-17–producing T cells. (A) Percentages of memory CD4+ T cells producing IL-17A, -22, -17F, and IFN-γ, as determined ex vivo by flow cytometry, after 12 h of stimulation with PMA and ionomycin. Horizontal lines indicate the mean value. (B) IL-17A and -22 production, measured by ELISA, in the supernatants of whole blood after 24 h of stimulation with PMA and ionomycin. Horizontal lines indicate the mean value. These two experiments were conducted in parallel in 30 healthy controls, 8 healthy relatives, and 7 patients from kindreds C, D, E, and H. *P < 0.05; **P < 0.005; ***P < 0.0005 (two-tailed Mann–Whitney test).
Fig. S7.

Response to bacteria and yeasts in whole blood. Production of IL-17A, -6, and IFN-γ, measured by ELISA, in whole blood from four controls (black circles), four patients (red circles), and three healthy relatives (black triangles) after 3 d of stimulation is shown. Horizontal bars represent the mean value. BCG, Bacille de Calmette et Guérin; HKCA, heat-killed C. albicans; HKSA, heat-killed S. aureus; HKSC, heat-killed S. cerevisiae; LPS, lipopolysaccharide; NS, not stimulated; VSV, vesicular stomatitis virus.
Discussion
We report complete AR IL-17RA deficiency in 21 patients from 12 unrelated kindreds and 6 ethnic groups (9). All 12 alleles, including the 2 alleles (p.D387N and p.Y591Sfs*29) encoding surface-expressed receptors, are loss-of-function in terms of responses to IL-17A, -17F, and -17A/F in fibroblasts. In addition, p.D387N is also loss-of-function for the response to IL-17E/IL-25 in PBMCs. Interestingly, the missense allele encodes a surface receptor in monocytes only. The clinical and cellular phenotypes of the two patients with this allele did not differ from those of patients with loss-of-expression alleles. This finding suggests that the p.D387N allele encodes a receptor that is present but not functional on monocytes, due to impairment of the SEFIR-mediated interaction with the adaptor ACT1. An alternative, but less likely, hypothesis is that IL-17RA–dependent signaling in monocytes may be redundant for mucocutaneous immunity to Candida. The cell-surface expression of dysfunctional receptors is the second genetic form of AR IL-17RA deficiency to be described. The detection of surface IL-17RA should not, therefore, exclude a diagnosis of IL-17RA deficiency, as previously shown for other cytokine receptors, such as IFN-γR1 (66–69), IFN-γR2 (70, 71), IL-12Rβ1 (72, 73), and IL-10RA (74, 75). IL-17RA deficiency has recently been reported in two siblings from Sri Lanka (60). These siblings are homozygous for a large chromosomal deletion, also encompassing CECR1 (encoding ADA2) and XKR3 (encoding X Kell blood group-related 3). These two patients displayed CMC and staphylococcal skin infections, together with a chronic inflammatory disease possibly related to ADA2 deficiency. Collectively, these clinical observations suggest that AR IL-17RA deficiency is the second most common known genetic etiology of CMCD, after GOF STAT1, and the most common known etiology of isolated, as opposed to syndromic, CMCD (51).
All of the IL17RA alleles tested were null, because the responses to IL-17A/IL-17F homodimers and heterodimers in the patients’ fibroblasts (cells tested displaying the best induction of IL-6 and GRO-α in controls)—and, by inference, probably in PBMCs (9)—and responses to IL-17E/IL-25 in their PBMCs were abolished. We predict that none of the cell types normally expressing IL-17RA in healthy individuals (whether hematopoietic or nonhematopoietic) respond to IL-17RA–dependent cytokines in patients. Nevertheless, the susceptibility to infection of IL-17RA–deficient patients appeared to be restricted to certain mucocutaneous barrier sites. In addition to CMC and cutaneous staphylococcal infections, several patients presented bacterial infections of the respiratory tract, which may not be coincidental (76). In a mouse model of Klebsiella pneumoniae infection, IL-17RA signaling has been shown to be critical for the optimal production of chemokines and granulocyte colony-stimulating factor in the lungs and for neutrophil recruitment and survival (77). The skin and mucosal phenotype of the patients may be accounted for, at least in part, by human keratinocytes and bronchial epithelial cells having a much greater dependence than other cell types on the synergistic effect of IL-17 cytokines (IL-17A and -17F in particular) and inflammatory cytokines (such as TNF-α) for the production of chemokines and antimicrobial peptides (78). It remains unclear how IL-17A, -A/F, and -F cooperate with other cytokines, but multiple ACT1-dependent mechanisms involving the promoter (as for IL-6) and/or mRNA stabilization (e.g., the GRO-α mRNA) (79, 80) are probably involved, as suggested by the similar clinical and functional impacts of human IL-17RA and ACT1 deficiencies (9, 11).
CMC is the key clinical presentation of patients with AR IL-17RA (refs. 9 and 60 and this work), AD IL-17F (9), AR ACT1 (11), or AR IL-17RC deficiency (10). Nevertheless, the heterogeneous phenotypes of these patients suggest a continuum of severity, ranging from a relatively mild phenotype in patients with IL-17F or -17RC deficiency to a more severe phenotype in patients with ACT1 or IL-17RA deficiency. Patients with ACT1 or IL-17RA deficiency are also susceptible to staphylococcal skin infections and bacterial respiratory infections, which tend to run a more chronic course. It is too early to draw firm conclusions, given the small number of patients identified. However, each of these genetic defects may have a different impact on IL-17 immunity. For example, in addition to acting in concert with IL-17RC for responses to IL-17A/F, IL-17RA acts with IL-17RB in mice (81) and with IL-17RE in mice and humans (82, 83) in responses to IL-17E/IL-25 and -17C, respectively. The function of IL-17RD is poorly defined and its ligand is unknown, but studies in mice have shown that ACT1 is essential for the signal transduction mediated by the individual IL-17RA (84, 85), IL-17RB (86), IL-17RC (87), and IL-17RE (88) subunits. Unlike those of IL-17RC–deficient patients, PBMCs from ACT1-deficient and IL-17RA–deficient patients do not respond to IL-17E/IL-25 (10, 11). The role of human IL-17E/IL-25 is unknown, in the absence of known patients bearing specific mutations, but its mouse counterpart is known to promote “Th2”-mediated responses (89, 90) and to be involved in immunity to parasitic infections (91–93). We were unable to detect cellular responses to IL-17B, -17D, and even -17C (83) in control fibroblasts, keratinocytes, or leukocytes. This finding precluded the testing of such responses in IL-17RA–deficient or other patients with CMC. Human IL-17C may play a redundant role in protective mucocutaneous immunity to Candida, because IL-17C– and IL-17RE–deficient mice clear Candida infections normally (94). The role of each human IL-17 cytokine in vivo will be determined from the description of patients bearing mutations, as reported for IL-17F (9). Overall, our data demonstrate that human signaling via IL-17RA (in response to at least IL-17A, -17A/F, -17F, and -17E/IL-25) is essential for mucocutaneous immunity to C. albicans and Staphylococcus. They also suggest that IL-17RA–dependent signaling is important for protective immunity to various bacteria in the respiratory tract.
Materials and Methods
Massively Parallel Sequencing.
Genomic DNA extracted from the peripheral blood cells of each patient was sheared with a Covaris S2 Ultrasonicator. An adaptor-ligated library was prepared with the Paired-End Sample Prep kit V1 (Illumina). Exome capture was performed with the SureSelect Human All Exon kit (Agilent Technologies). Single-end sequencing was performed on an Illumina Genome Analyzer IIx (Illumina), generating 72-base reads.
Molecular Genetics.
GenomicDNA was isolated from whole blood by a phenol/chloroform extraction method. IL17RA gDNA was amplified with specific primers (PCR amplification conditions and primer sequences are available in Table S1). PCR products were analyzed by electrophoresis in 1% agarose gels, sequenced with the Big Dye Terminator cycle sequencing kit (Applied Biosystems), and analyzed on an ABI Prism 3700 (Applied Biosystems).
Table S1.
PCR primers and conditions for IL17RA genomic DNA amplification
| Exon | Forward primer | Reverse primer |
| Exon 1 | GTCCCAGACTAAACTCCTCC | GCAGCATCCTGGCGGCTAC |
| Exon 2 | CTGAGAATGAGCCAGTGGAG | GGCATGGCAAGACTTCTGTC |
| Exon 3 | CTGAGCTGTTTGCTGTCTAGC | CCAATCCTGAGCCTGACTGG |
| Exon 4 | GACACACCCAGCACTTGTC | GAAGTCTGTACTGGTGTCATC |
| Exon 5 | GCATAGATGGGTGACAGAGGTG | GGTGCTTCCTTCTCACAGAGG |
| Exon 6 and 7 | CAGATTTCTGAAGGCAGAGGC | GCAACGCGGTTCTGTCAGAC |
| Exon 8 | CTCTGGACAGCCTCTCCATG | CTCCTGTCCACTCTATAATTGC |
| Exon 9 | GCTGCTGGCTGGAAGGCATG | CAGGTAAGGACGCAGCACAG |
| Exon 10 and 11 | CTGGGAAGGGTTAAGAATGC | GGTGCTGGTATTAGAGCCTG |
| Exon 12 | GTGCGACCACCTAGCACG | GGGAGTTAGAGCACAGGAG |
| Exon 13-a | CTCCTGGGCTGGCAGGCAC | CGTCACAGCTGACCTCGCTG |
| Exon 13-b | CAGCCATGAACATGATCCTCC | CTTCGAGGTGCTCCCTCAC |
| Exon 13-c | GCAGCAGTGGCAAAGCTGG | CAGATGAATACGTGCACACAC |
| Exon 13-d | GGACTCACGGAAATGGAGGAAGAG | CGTTTACATCCCGCGTGACCATC |
Conditions: 2.5 mM MgCl2; 7.5% (vol/vol) DMSO; melting temperature: 59 °C; 1 min, 1 min, 1 min × 38 cycles.
Cell Activation.
For the ex vivo evaluation of IL-17A– and IL-22–producing T cells by ELISA, we used 250 µL of whole blood diluted in RPMI (500 µL final volume) to seed 48-well plates. We added 40 ng/mL PMA and 10−5 M ionomycin and incubated the plates for 24 h. The supernatants were then collected for ELISA (R&D Systems). For the evaluation of the response to IL-17E/IL-25, fresh PBMCs were cultured in the presence of 100 ng/mL thymic stromal lymphopoietin (R&D Systems; 1398-TS-010/CF0) in X-VIVO 15 (Lonza) plus 5% human AB serum (Lonza) for 24 h. PBMCs were collected, washed, and resuspended at a density of 4 × 106 cells per well in 48-well plates, in a final volume of 0.5 mL per well, in the presence of 10 ng/mL recombinant human IL-2 (R&D Systems) and 10 ng/mL recombinant human IL-17E (R&D Systems). After 3 d, IL-5 secretion was assessed by ELISA (DY205; R&D Systems). SV40-transformed fibroblasts were plated in 48-well plates at a density of 100,000 cells per well in 0.5 mL of DMEM/10% (vol/vol) FBS. They were incubated for 24 h and then left unstimulated or stimulated for 24 h with recombinant human IL-17A, -17F, and -17A/F (100 ng/mL), with or without TNF-α (20 ng/mL) purchased from R&D Systems. The supernatants were collected for ELISA for IL-6 (Sanquin) and GRO-α (R&D Systems), carried out in accordance with the kit manufacturer’s instructions.
Flow Cytometry.
For the ex vivo evaluation of IL-17A–, IL-17F–, IL-22– and IFN-γ–producing T cells by flow cytometry, PBMCs were dispensed into 48-well plates at a density of 3 × 106 cells per mL in RPMI/10% (vol/vol) FBS for 12 h with 40 ng/mL PMA plus 10−5 M ionomycin, in the presence of a secretion inhibitor (1 µl/mL GolgiPlug; BD Biosciences). The cells were washed and surface-labeled with PE-Cy7 mouse anti-human CD3 (SK7; BD Biosciences), CD4-APC-Vio770, human (M-T321; Miltenyi Biotec), Brilliant Violet 421 anti-mouse CD197 (CCR7) (G043H7; BioLegend), PE-CF594 mouse anti-human CD45RA (HI100; BD Biosciences), and LIVE/DEAD Fixable Aqua Dead Cell (L34957; Thermo Fisher) in PBS/2% (vol/vol) FBS/2 mM EDTA for 20 min on ice. Cells were then washed twice with PBS/2% (vol/vol) FBS/2 mM EDTA, fixed by incubation with 100 µL of BD Cytofix for 30 min on ice, and washed twice with BD Cytoperm (Cytofix/Cytoperm Plus, fixation/permeabilization kit; BD Biosciences). Cells were then incubated for 1 h on ice with antibodies purchased from Ebiosciences—anti-human IL-17A Alexa Fluor 488 (eBio64DEC17), anti-human IL-17F PE (SHLR17), anti-human/mouse IL-22 APC (IL22JOP), and anti-human IFN gamma Alexa Fluor 700 (4S.B3) —washed twice with Cytoperm, and analyzed with a FACS Gallios flow cytometer. For the evaluation of IL-17RA expression, SV40-transformed fibroblasts or PBMCs were labeled simultaneously with LIVE/DEAD Fixable Aqua Dead Cell, Alexa Fluor 647-mouse IgG1 isotype control antibodies (MOPC-21; BioLegend) or Alexa Fluor 647–anti-human IL-17AR (BG/hIL17AR; BioLegend). PBMCs were also labeled with CD14-PE-Vio770, human (TÜK4), or CD3-VioBlue, human (BW264/56), purchased from Miltenyi Biotec; APC-Cy7 mouse anti-human CD19 (SJ25C1) and PE-CF594 mouse anti-human CD56 (B159) purchased from BD Biosciences, and analyzed by flow cytometry in a FACS Gallios flow cytometer.
Cell Complementation.
IL-17RA–deficient SV40-transformed fibroblasts were transfected with either empty pORF9-msc vectors or with the pORF9-hIL17RA vector encoding the wild-type human IL-17RA (Cayla-InvivoGen), with the Lipofectamine LTX transfection kit (Invitrogen), according to the manufacturer’s instructions. At 24 h later, cells were stimulated with IL-17A (100 ng/mL), with or without TNF-α (20 ng/mL), for a further 24 h. The supernatants were collected for the assessment of IL-6 and GRO-α levels by ELISA, and the cells were collected for the evaluation of IL-17RA expression by FACS analysis.
Full-Length RT-PCR for DEFB4A and Taqman Probe Detection.
Total RNA was extracted with the RNeasy minikit (Qiagen) and reverse-transcribed to generate cDNA, with the High Capacity cDNA Reverse Transcription Kit (4368813; Invitrogen). Taqman probes for DEFB4A (Hs00823638_m1; Invitrogen) were used to detect mRNA synthesis, with normalization on the basis of GUS expression (Human GUSB Endogenous Control VIC/MGB Probe; 4326320E; Primer Limited; Invitrogen).
Healthy Controls.
The healthy controls were volunteer blood donors of European and Turkish origin.
Study Approval.
The experiments described here were conducted in accordance with local, national, and international regulations and were approved by the French Ethics committee (CPP Ile-de-France 2, ID-RCB: 2010-A00636-33), French National Agency for Medicines and Health Products Safety (B100712-40), and the French Ministry of Research (IE-2010-547). Informed consent was obtained from all patients or their families, in the case of minors, in accordance with World Medical Association rules, the Helsinki Declaration, and European Union directives.
SI Materials and Methods
Cell Purification and Culture.
Human PBMCs were isolated by Ficoll–Hypaque density gradient centrifugation (Amersham Pharmacia Biotech) from whole blood. Primary human fibroblasts, obtained from skin biopsy specimens, were immortalized with SV-40T antigen (SV-40 fibroblasts) and cultured in DMEM (Gibco BRL, Invitrogen) supplemented with 10% (vol/vol) FBS (Gibco BRL, Invitrogen).
Immunophenotyping.
For the detailed phenotyping of lymphocyte subsets, CD4+ and CD8+ T cells, CD4+ effector T cells, B cells, natural killer (NK) cells, regulatory T cells, and fresh PBMCs were labeled with BV786 mouse anti-human CD3 (SK7), PE mouse anti-human CD4 (RPA-T4), PE-CF594 mouse anti-human CD45RA (HI100), FITC mouse anti-human CD27 (L128), BV650 mouse anti-human CD8 (RPA-T8), BV711 mouse anti-human CD161 (DX12), PE-CF594 mouse anti-human CD56 (B159), BV421 mouse anti-human CD25 (M-A251), BV711 mouse anti-human CD4 (SK3), BV711 mouse anti-human CD21 (B-ly4), APC-Cy7 mouse anti-human CD19 (SJ25C1), and Alexa Fluor 488 mouse anti-human FoxP3 (259D/C7) antibodies purchased from BD Biosciences; anti-human CD20-FITC (LT20), anti-human IgM-PE (PJ2-22H3), anti-human IgD-PE (IgD26), anti-human CD38-PE-Vio770 (REA572), anti-human CD183 (CXCR3)-VioBright FITC (REA232), anti-human CD196 (CCR6)-PE (REA190), anti-human CD194 (CCR4)-PE (REA279), anti-human CD185 (CXCR5)-PE-Vio770 (REA103), anti-human CD4-APC-Vio770 (M-T321), anti-human CD3-PE-Vio770 (BW264/56), anti-human CD159a (NKG2A)-APC (REA110), anti-human CD159c (NKG2C)-PE (REA205), anti-human TCR-Vα7.2-APC-Vio770 (REA179), and anti-human TCRγ/δ-VioBlue (11F2) antibodies purchased from Miltenyi Biotec; CD158a,h-PE IOTest (EB6B) and CD158b1/b2,j (GL183) antibodies purchased from Beckman Coulter; and Brilliant Violet 421 anti-human CD197 (CCR7) antibody (G043H7), and Brilliant Violet 421 anti-human CD27 antibody (M-T271) purchased from BioLegend.
Transfection and Coimmunoprecipitation.
HEK293 cells were transiently transfected with the indicated plasmids in the presence of Lipofectamine 2000 (Thermo Fisher Scientific) before harvest for coimmunoprecipitation. After 48 h of transfection, cells were lysed in lysis buffer [1% Triton X-100, 20 mM Hepes (pH 7.4), 150 mM NaCl, 12.5 mM β-glycerophosphate, 1.5 mM MgCl2, 10 mM NaF, 2 mM DTT, 1 mM sodium orthovanadate, and 2 mM EGTA] supplemented with protease inhibitor mixture (Complete; Roche). Cell extracts were incubated with antibody (1 µg) and protein A beads (20 µL). After overnight incubation, the beads were washed four times with lysis buffer and then mixed with 40 μL of Laemmli buffer. The immunoprecipitates were resolved by SDS/PAGE and analyzed by Western blotting.
Acknowledgments
We thank the members of the laboratory for helpful discussions; Yelena Nemiroskaya, Eric Anderson, Martine Courat, and Lahouari Amar for expert secretarial assistance; and Maya Chrabieh, Lazaro Lorenzo, and Tiffany Nivare for technical assistance. This work was supported by French National Research Agency (ANR) Grants ANR GENCMCD-11-BSV3–005-01 (to A.P.), ANR EURO-CMC-14-RARE-0005-02), and “Investissement d’avenir” Program Grant ANR-10-IAHU-01; Laboratoire d’Excellence Integrative Biology of Emerging Infectious Diseases Grant ANR-10-LABX-62-IBEID; the Jeffrey Modell Centers Network; the St. Giles Foundation; the Rockefeller University; INSERM; and Paris Descartes University. This study was supported in part by Japan Society for the Promotion of Science Grants in Aid for Scientific Research 16H05355 and 16K1552 and the Practical Research Project for Rare/Intractable Diseases of the Japan Agency for Medical Research and Development, AMED. R.L. was supported by the SNFMI (Bourse Marcel Simon), the INSERM PhD program for medical doctors (poste d’accueil INSERM), and a Fulbright grant (Franco-American commission). V.B. was supported by ANR Grant NKIR-ANR-13-PDOC-0025-01. K.M. was supported by a Japan Foundation for Pediatric Research fellowship grant. L.Y.A.C. was supported by the Singapore National Medical Research Council and National University Health System. F.G. is a scholar of the Else Kröner Fresenius Clinical Research School München “Rare diseases of the immune system—from pathophysiology to new therapies” (Grant 2013_Kolleg.18).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1618300114/-/DCSupplemental.
References
- 1.Puel A, et al. Inborn errors of human IL-17 immunity underlie chronic mucocutaneous candidiasis. Curr Opin Allergy Clin Immunol. 2012;12(6):616–622. doi: 10.1097/ACI.0b013e328358cc0b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chandesris M-O, et al. Autosomal dominant STAT3 deficiency and hyper-IgE syndrome: Molecular, cellular, and clinical features from a French national survey. Medicine (Baltimore) 2012;91(4):e1–e19. doi: 10.1097/MD.0b013e31825f95b9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kisand K, Peterson P. Autoimmune polyendocrinopathy candidiasis ectodermal dystrophy. J Clin Immunol. 2015;35(5):463–478. doi: 10.1007/s10875-015-0176-y. [DOI] [PubMed] [Google Scholar]
- 4.Wells RS, Higgs JM, Macdonald A, Valdimarsson H, Holt PJ. Familial chronic muco-cutaneous candidiasis. J Med Genet. 1972;9(3):302–310. doi: 10.1136/jmg.9.3.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chilgren RA, Quie PG, Meuwissen HJ, Hong R. Chronic mucocutaneous candidiasis, deficiency of delayed hypersensitivity, and selective local antibody defect. Lancet. 1967;2(7518):688–693. doi: 10.1016/s0140-6736(67)90974-9. [DOI] [PubMed] [Google Scholar]
- 6.Puel A, et al. Inborn errors of mucocutaneous immunity to Candida albicans in humans: A role for IL-17 cytokines? Curr Opin Immunol. 2010;22(4):467–474. doi: 10.1016/j.coi.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Casanova J-L. Human genetic basis of interindividual variability in the course of infection. Proc Natl Acad Sci USA. 2015;112(51):E7118–E7127. doi: 10.1073/pnas.1521644112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Casanova J-L. Severe infectious diseases of childhood as monogenic inborn errors of immunity. Proc Natl Acad Sci USA. 2015;112(51):E7128–E7137. doi: 10.1073/pnas.1521651112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Puel A, et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science. 2011;332(6025):65–68. doi: 10.1126/science.1200439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ling Y, et al. Inherited IL-17RC deficiency in patients with chronic mucocutaneous candidiasis. J Exp Med. 2015;212(5):619–631. doi: 10.1084/jem.20141065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boisson B, et al. An ACT1 mutation selectively abolishes interleukin-17 responses in humans with chronic mucocutaneous candidiasis. Immunity. 2013;39(4):676–686. doi: 10.1016/j.immuni.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol. 2009;9(8):556–567. doi: 10.1038/nri2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aldave JC, et al. A 1-year-old girl with a gain-of-function STAT1 mutation treated with hematopoietic stem cell transplantation. J Clin Immunol. 2013;33(8):1273–1275. doi: 10.1007/s10875-013-9947-5. [DOI] [PubMed] [Google Scholar]
- 14.Altman MC, et al. A young boy with a novel, autosomal-dominant signal transducer and activator of transcription 1 (STAT1) hypermorphic mutation presenting with pneumocystis Jirovecii pneumonia (PJP), chronic mucocutaneous candidiasis (CMC), and combined immunodeficiency. J Allergy Clin Immunol. 2014;133(2 Suppl):AB250. [Google Scholar]
- 15.Baer Ellington AE, Shih JA. Sporadic case of chronic mucocutaneous candidiasis (CMC) due to a gain-of-function mutation in STAT1 in a 13 year old female. J Allergy Clin Immunol. 2014;133(2 Suppl):AB250. [Google Scholar]
- 16.Bina SM, et al. Variable presentations of gain of function STAT1 mutations within a single institution with features beyond chronic mucocutaneous candidiasis. J Allergy Clin Immunol. 2014;135(2 Suppl):AB186. [Google Scholar]
- 17.Depner M, et al. The extended clinical phenotype of 26 patients with chronic mucocutaneous candidiasis due to gain-of-function mutations in STAT1. J Clin Immunol. 2016;36(1):73–84. doi: 10.1007/s10875-015-0214-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dhalla F, et al. Chronic mucocutaneous candidiasis: Characterization of a family with STAT-1 gain-of-function and development of an ex-vivo assay for Th17 deficiency of diagnostic utility. Clin Exp Immunol. 2016;184(2):216–227. doi: 10.1111/cei.12746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dotta L, et al. Clinical heterogeneity of dominant chronic mucocutaneous candidiasis disease: Presenting as treatment-resistant candidiasis and chronic lung disease. Clin Immunol. 2016;164:1–9. doi: 10.1016/j.clim.2015.12.010. [DOI] [PubMed] [Google Scholar]
- 20.Frans G, et al. Gain-of-function mutations in signal transducer and activator of transcription 1 (STAT1): Chronic mucocutaneous candidiasis accompanied by enamel defects and delayed dental shedding. J Allergy Clin Immunol. 2014;134(5):1209–13.e6. doi: 10.1016/j.jaci.2014.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Giardino G, et al. Novel STAT1 gain-of-function mutation and suppurative infections. Pediatr Allergy Immunol. 2016;27(2):220–223. doi: 10.1111/pai.12496. [DOI] [PubMed] [Google Scholar]
- 22.Higgins E, et al. Use of ruxolitinib to successfully treat chronic mucocutaneous candidiasis caused by gain-of-function signal transducer and activator of transcription 1 (STAT1) mutation. J Allergy Clin Immunol. 2015;135(2):551–553. doi: 10.1016/j.jaci.2014.12.1867. [DOI] [PubMed] [Google Scholar]
- 23.Hori T, et al. Autosomal-dominant chronic mucocutaneous candidiasis with STAT1-mutation can be complicated with chronic active hepatitis and hypothyroidism. J Clin Immunol. 2012;32(6):1213–1220. doi: 10.1007/s10875-012-9744-6. [DOI] [PubMed] [Google Scholar]
- 24.Kataoka S, et al. Extrapulmonary tuberculosis mimicking Mendelian susceptibility to mycobacterial disease in a patient with signal transducer and activator of transcription 1 (STAT1) gain-of-function mutation. J Allergy Clin Immunol. 2016;137(2):619–622.e1. doi: 10.1016/j.jaci.2015.06.028. [DOI] [PubMed] [Google Scholar]
- 25.Kilic SS, Puel A, Casanova J-L. Orf Infection in a patient with Stat1 gain-of-function. J Clin Immunol. 2015;35(1):80–83. doi: 10.1007/s10875-014-0111-7. [DOI] [PubMed] [Google Scholar]
- 26.Kumar N, et al. Gain-of-function signal transducer and activator of transcription 1 (STAT1) mutation-related primary immunodeficiency is associated with disseminated mucormycosis. J Allergy Clin Immunol. 2014;134(1):236–239. doi: 10.1016/j.jaci.2014.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee PPW, et al. Penicillium marneffei infection and impaired IFN-γ immunity in humans with autosomal-dominant gain-of-phosphorylation STAT1 mutations. J Allergy Clin Immunol. 2014;133(3):894–6.e5. doi: 10.1016/j.jaci.2013.08.051. [DOI] [PubMed] [Google Scholar]
- 28.Liu L, et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med. 2011;208(8):1635–1648. doi: 10.1084/jem.20110958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martinez-Martinez L, et al. A novel gain-of-function STAT1 mutation resulting in basal phosphorylation of STAT1 and increased distal IFN-γ-mediated responses in chronic mucocutaneous candidiasis. Mol Immunol. 2015;68(2 Pt C):597–605. doi: 10.1016/j.molimm.2015.09.014. [DOI] [PubMed] [Google Scholar]
- 30.Mekki N, et al. IL-17 T cells’ defective differentiation in vitro despite normal range ex vivo in chronic mucocutaneous candidiasis due to STAT1 mutation. J Invest Dermatol. 2014;134(4):1155–1157. doi: 10.1038/jid.2013.480. [DOI] [PubMed] [Google Scholar]
- 31.Mizoguchi Y, et al. Simple diagnosis of STAT1 gain-of-function alleles in patients with chronic mucocutaneous candidiasis. J Leukoc Biol. 2014;95(4):667–676. doi: 10.1189/jlb.0513250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mössner R, et al. Ruxolitinib induces interleukin 17 and ameliorates chronic mucocutaneous candidiasis caused by STAT1 gain-of-function mutation. Clin Infect Dis. 2016;62(7):951–953. doi: 10.1093/cid/ciw020. [DOI] [PubMed] [Google Scholar]
- 33.Romberg N, et al. Gain-of-function STAT1 mutations are associated with PD-L1 overexpression and a defect in B-cell survival. J Allergy Clin Immunol. 2013;131(6):1691–1693. doi: 10.1016/j.jaci.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruda Wessell KM, et al. A young adult male with chronic mucocutaneous candidiasis (CMC) with signal transduction and activator of transcription 1 (STAT 1) mutation and progressive multifocal leukoencephalopathy (PML) J Allergy Clin Immunol. 2015;135(2 Suppl):AB186. [Google Scholar]
- 35.Al Rushood M, et al. Autosomal dominant cases of chronic mucocutaneous candidiasis segregates with mutations of signal transducer and activator of transcription 1, but not of Toll-like receptor 3. J Pediatr. 2013;163(1):277–279. doi: 10.1016/j.jpeds.2013.02.040. [DOI] [PubMed] [Google Scholar]
- 36.Nielsen J, et al. A STAT1-gain-of-function mutation causing Th17 deficiency with chronic mucocutaneous candidiasis, psoriasiform hyperkeratosis and dermatophytosis. BMJ Case Rep. 2015;2015:bcr2015211372. doi: 10.1136/bcr-2015-211372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Salim N, Leiding J. Fungal granuloma and chronic mucocutaneous candidiasis due to autosomal dominant gain of function STAT1 mutation. J Allergy Clin Immunol. 2014;133(2 Suppl):AB251. [Google Scholar]
- 38.Sampaio EP, et al. Signal transducer and activator of transcription 1 (STAT1) gain-of-function mutations and disseminated coccidioidomycosis and histoplasmosis. J Allergy Clin Immunol. 2013;131(6):1624–1634. doi: 10.1016/j.jaci.2013.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sharfe N, et al. Fatal combined immunodeficiency associated with heterozygous mutation in STAT1. J Allergy Clin Immunol. 2014;133(3):807–817. doi: 10.1016/j.jaci.2013.09.032. [DOI] [PubMed] [Google Scholar]
- 40.Smeekens SP, et al. STAT1 hyperphosphorylation and defective IL12R/IL23R signaling underlie defective immunity in autosomal dominant chronic mucocutaneous candidiasis. PLoS One. 2011;6(12):e29248. doi: 10.1371/journal.pone.0029248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Soltész B, et al. New and recurrent gain-of-function STAT1 mutations in patients with chronic mucocutaneous candidiasis from Eastern and Central Europe. J Med Genet. 2013;50(9):567–578. doi: 10.1136/jmedgenet-2013-101570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takezaki S, et al. Chronic mucocutaneous candidiasis caused by a gain-of-function mutation in the STAT1 DNA-binding domain. J Immunol. 2012;189(3):1521–1526. doi: 10.4049/jimmunol.1200926. [DOI] [PubMed] [Google Scholar]
- 43.Tanimura M, et al. Recurrent inflammatory aortic aneurysms in chronic mucocutaneous candidiasis with a gain-of-function STAT1 mutation. Int J Cardiol. 2015;196:88–90. doi: 10.1016/j.ijcard.2015.05.183. [DOI] [PubMed] [Google Scholar]
- 44.Tóth B, et al. Herpes in STAT1 gain-of-function mutation. Lancet. 2012;379(9835):2500, and erratum (2012) 380(9844):806. doi: 10.1016/S0140-6736(12)60365-1. [DOI] [PubMed] [Google Scholar]
- 45.Uzel G, et al. Dominant gain-of-function STAT1 mutations in FOXP3 wild-type immune dysregulation-polyendocrinopathy-enteropathy-X-linked-like syndrome. J Allergy Clin Immunol. 2013;131(6):1611–1623. doi: 10.1016/j.jaci.2012.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van de Veerdonk FL, et al. STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N Engl J Med. 2011;365(1):54–61. doi: 10.1056/NEJMoa1100102. [DOI] [PubMed] [Google Scholar]
- 47.Wang X, et al. Exome sequencing reveals a signal transducer and activator of transcription 1 (STAT1) mutation in a child with recalcitrant cutaneous fusariosis. J Allergy Clin Immunol. 2013;131(4):1242–1243. doi: 10.1016/j.jaci.2012.11.005. [DOI] [PubMed] [Google Scholar]
- 48.Wildbaum G, et al. Continuous G-CSF therapy for isolated chronic mucocutaneous candidiasis: Complete clinical remission with restoration of IL-17 secretion. J Allergy Clin Immunol. 2013;132(3):761–764. doi: 10.1016/j.jaci.2013.04.018. [DOI] [PubMed] [Google Scholar]
- 49.Yamazaki Y, et al. Two novel gain-of-function mutations of STAT1 responsible for chronic mucocutaneous candidiasis disease: Impaired production of IL-17A and IL-22, and the presence of anti-IL-17F autoantibody. J Immunol. 2014;193(10):4880–4887. doi: 10.4049/jimmunol.1401467. [DOI] [PubMed] [Google Scholar]
- 50.Zerbe CS, et al. Progressive multifocal leukoencephalopathy in primary immune deficiencies: Stat1 gain of function and review of the literature. Clin Infect Dis. 2016;62(8):986–994. doi: 10.1093/cid/civ1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Toubiana J, et al. International STAT1 Gain-of-Function Study Group Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood. 2016;127(25):3154–3164. doi: 10.1182/blood-2015-11-679902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zheng J, et al. Gain-of-function STAT1 mutations impair STAT3 activity in patients with chronic mucocutaneous candidiasis (CMC) Eur J Immunol. 2015;45(10):2834–2846. doi: 10.1002/eji.201445344. [DOI] [PubMed] [Google Scholar]
- 53.Okada S, et al. IMMUNODEFICIENCIES. Impairment of immunity to Candida and Mycobacterium in humans with bi-allelic RORC mutations. Science. 2015;349(6248):606–613. doi: 10.1126/science.aaa4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.de Beaucoudrey L, et al. Mutations in STAT3 and IL12RB1 impair the development of human IL-17-producing T cells. J Exp Med. 2008;205(7):1543–1550. doi: 10.1084/jem.20080321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ma CS, et al. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J Exp Med. 2008;205(7):1551–1557. doi: 10.1084/jem.20080218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Milner JD, et al. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature. 2008;452(7188):773–776. doi: 10.1038/nature06764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Renner ED, et al. Novel signal transducer and activator of transcription 3 (STAT3) mutations, reduced T(H)17 cell numbers, and variably defective STAT3 phosphorylation in hyper-IgE syndrome. J Allergy Clin Immunol. 2008;122(1):181–187. doi: 10.1016/j.jaci.2008.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kisand K, et al. Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. J Exp Med. 2010;207(2):299–308. doi: 10.1084/jem.20091669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Puel A, et al. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J Exp Med. 2010;207(2):291–297. doi: 10.1084/jem.20091983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fellmann F, et al. IL-17 receptor A and adenosine deaminase 2 deficiency in siblings with recurrent infections and chronic inflammation. J Allergy Clin Immunol. 2016;137(4):1189–1196.e1–2. doi: 10.1016/j.jaci.2015.07.053. [DOI] [PubMed] [Google Scholar]
- 61.Onishi RM, et al. SEF/IL-17R (SEFIR) is not enough: An extended SEFIR domain is required for il-17RA-mediated signal transduction. J Biol Chem. 2010;285(43):32751–32759. doi: 10.1074/jbc.M110.121418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Maitra A, et al. Distinct functional motifs within the IL-17 receptor regulate signal transduction and target gene expression. Proc Natl Acad Sci USA. 2007;104(18):7506–7511. doi: 10.1073/pnas.0611589104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Scott EM, et al. Greater Middle East Variome Consortium Characterization of Greater Middle Eastern genetic variation for enhanced disease gene discovery. Nat Genet. 2016;48(9):1071–1076. doi: 10.1038/ng.3592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Itan Y, et al. The mutation significance cutoff: Gene-level thresholds for variant predictions. Nat Methods. 2016;13(2):109–110. doi: 10.1038/nmeth.3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kircher M, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Allende LM, et al. A point mutation in a domain of gamma interferon receptor 1 provokes severe immunodeficiency. Clin Diagn Lab Immunol. 2001;8(1):133–137. doi: 10.1128/CDLI.8.1.133-137.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jouanguy E, et al. In a novel form of IFN-gamma receptor 1 deficiency, cell surface receptors fail to bind IFN-gamma. J Clin Invest. 2000;105(10):1429–1436. doi: 10.1172/JCI9166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Camcioglu Y, et al. HHV-8-associated Kaposi sarcoma in a child with IFNgammaR1 deficiency. J Pediatr. 2004;144(4):519–523. doi: 10.1016/j.jpeds.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 69.Dorman SE, et al. Clinical features of dominant and recessive interferon gamma receptor 1 deficiencies. Lancet. 2004;364(9451):2113–2121. doi: 10.1016/S0140-6736(04)17552-1. [DOI] [PubMed] [Google Scholar]
- 70.Vogt G, et al. Gains of glycosylation comprise an unexpectedly large group of pathogenic mutations. Nat Genet. 2005;37(7):692–700. doi: 10.1038/ng1581. [DOI] [PubMed] [Google Scholar]
- 71.Vogt G, et al. Complementation of a pathogenic IFNGR2 misfolding mutation with modifiers of N-glycosylation. J Exp Med. 2008;205(8):1729–1737. doi: 10.1084/jem.20071987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fieschi C, et al. A novel form of complete IL-12/IL-23 receptor beta1 deficiency with cell surface-expressed nonfunctional receptors. Blood. 2004;104(7):2095–2101. doi: 10.1182/blood-2004-02-0584. [DOI] [PubMed] [Google Scholar]
- 73.Fieschi C, et al. Low penetrance, broad resistance, and favorable outcome of interleukin 12 receptor beta1 deficiency: Medical and immunological implications. J Exp Med. 2003;197(4):527–535. doi: 10.1084/jem.20021769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mao H, et al. Exome sequencing identifies novel compound heterozygous mutations of IL-10 receptor 1 in neonatal-onset Crohn’s disease. Genes Immun. 2012;13(5):437–442. doi: 10.1038/gene.2012.8. [DOI] [PubMed] [Google Scholar]
- 75.Begue B, et al. Defective IL10 signaling defining a subgroup of patients with inflammatory bowel disease. Am J Gastroenterol. 2011;106(8):1544–1555. doi: 10.1038/ajg.2011.112. [DOI] [PubMed] [Google Scholar]
- 76.McAleer JP, Kolls JK. Directing traffic: IL-17 and IL-22 coordinate pulmonary immune defense. Immunol Rev. 2014;260(1):129–144. doi: 10.1111/imr.12183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ye P, et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med. 2001;194(4):519–527. doi: 10.1084/jem.194.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Minegishi Y, et al. Molecular explanation for the contradiction between systemic Th17 defect and localized bacterial infection in hyper-IgE syndrome. J Exp Med. 2009;206(6):1291–1301. doi: 10.1084/jem.20082767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shen F, Gaffen SL. Structure-function relationships in the IL-17 receptor: Implications for signal transduction and therapy. Cytokine. 2008;41(2):92–104. doi: 10.1016/j.cyto.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gaffen SL, Jain R, Garg AV, Cua DJ. The IL-23-IL-17 immune axis: From mechanisms to therapeutic testing. Nat Rev Immunol. 2014;14(9):585–600. doi: 10.1038/nri3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rickel EA, et al. Identification of functional roles for both IL-17RB and IL-17RA in mediating IL-25-induced activities. J Immunol. 2008;181(6):4299–4310. doi: 10.4049/jimmunol.181.6.4299. [DOI] [PubMed] [Google Scholar]
- 82.Song X, et al. IL-17RE is the functional receptor for IL-17C and mediates mucosal immunity to infection with intestinal pathogens. Nat Immunol. 2011;12(12):1151–1158. doi: 10.1038/ni.2155. [DOI] [PubMed] [Google Scholar]
- 83.Ramirez-Carrozzi V, et al. IL-17C regulates the innate immune function of epithelial cells in an autocrine manner. Nat Immunol. 2011;12(12):1159–1166. doi: 10.1038/ni.2156. [DOI] [PubMed] [Google Scholar]
- 84.Qian Y, et al. The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat Immunol. 2007;8(3):247–256. doi: 10.1038/ni1439. [DOI] [PubMed] [Google Scholar]
- 85.Chang SH, Park H, Dong C. Act1 adaptor protein is an immediate and essential signaling component of interleukin-17 receptor. J Biol Chem. 2006;281(47):35603–35607. doi: 10.1074/jbc.C600256200. [DOI] [PubMed] [Google Scholar]
- 86.Swaidani S, et al. The critical role of epithelial-derived Act1 in IL-17- and IL-25-mediated pulmonary inflammation. J Immunol. 2009;182(3):1631–1640. doi: 10.4049/jimmunol.182.3.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ho AW, et al. IL-17RC is required for immune signaling via an extended SEF/IL-17R signaling domain in the cytoplasmic tail. J Immunol. 2010;185(2):1063–1070. doi: 10.4049/jimmunol.0903739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chang SH, et al. Interleukin-17C promotes Th17 cell responses and autoimmune disease via interleukin-17 receptor E. Immunity. 2011;35(4):611–621. doi: 10.1016/j.immuni.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Angkasekwinai P, et al. Interleukin 25 promotes the initiation of proallergic type 2 responses. J Exp Med. 2007;204(7):1509–1517. doi: 10.1084/jem.20061675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fort MM, et al. IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity. 2001;15(6):985–995. doi: 10.1016/s1074-7613(01)00243-6. [DOI] [PubMed] [Google Scholar]
- 91.Angkasekwinai P, et al. Interleukin-25 (IL-25) promotes efficient protective immunity against Trichinella spiralis infection by enhancing the antigen-specific IL-9 response. Infect Immun. 2013;81(10):3731–3741. doi: 10.1128/IAI.00646-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhao A, et al. Critical role of IL-25 in nematode infection-induced alterations in intestinal function. J Immunol. 2010;185(11):6921–6929. doi: 10.4049/jimmunol.1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fallon PG, et al. Identification of an interleukin (IL)-25-dependent cell population that provides IL-4, IL-5, and IL-13 at the onset of helminth expulsion. J Exp Med. 2006;203(4):1105–1116. doi: 10.1084/jem.20051615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Conti HR, et al. Signaling through IL-17C/IL-17RE is dispensable for immunity to systemic, oral and cutaneous candidiasis. PLoS One. 2015;10(4):e0122807. doi: 10.1371/journal.pone.0122807. [DOI] [PMC free article] [PubMed] [Google Scholar]