

Abstract
Patients with inborn errors of IL-17F or IL-17RA display chronic mucocutaneous candidiasis (CMC). We report a biallelic missense mutation (T536I) in the adaptor molecule ACT1 in two siblings with CMC. The mutation, located in the SEFIR domain, abolished the homotypic interaction of ACT1 with IL-17 receptors, with no effect on homodimerization. The patients’ fibroblasts failed to respond to IL-17A and IL-17F, and their T cells to IL-17E. By contrast, healthy individuals homozygous for the common variant D10N, located in the ACT1 TNF receptor-associated factor (TRAF)-interacting domain and previously associated with psoriasis, had impaired, but not abolished, responses to IL-17 cytokines. SEFIR-independent interactions of ACT1 with other proteins, such as CD40, heat shock protein (HSP)70 and HSP90, were not affected by the T536I mutation. Overall, human IL-17A and IL-17F depend on ACT1 to mediate protective mucocutaneous immunity. Moreover, other ACT1-dependent IL-17 cytokines seem to be largely redundant in host defense.
Introduction
Chronic mucocutaneous candidiasis (CMC) is characterized by recurrent or persistent infections of the skin, nails, oral and genital mucosae with Candida albicans, and sometimes by staphylococcal skin infections (Glocker and Grimbacher, 2010; Lilic, 2012; Puel et al., 2012). Patients with inherited or acquired T cell immunodeficiencies often suffer from CMC. Patients with autosomal dominant (AD) hyper-IgE syndrome (HIES) due to heterozygous loss-of-function (LOF) mutations in STAT3 display CMC and a deficit of IL-17-producing T cells (Chandesris et al., 2012; de Beaucoudrey et al., 2008; Ma et al., 2008; Milner et al., 2008; Minegishi et al., 2009; Renner et al., 2008). Biallelic mutations of IL12B or IL12RB1 in patients with Mendelian susceptibility to mycobacterial disease (MSMD) can also lead to mild CMC, due to low proportions of IL-17-producing circulating T cells (de Beaucoudrey et al., 2008; de Beaucoudrey et al., 2010; Monia et al., 2013; Prando et al., 2013). Some patients with autosomal recessive (AR) CARD9 deficiency and invasive fungal diseases also have CMC and low proportions of circulating IL-17 T cells (Drewniak et al., 2013; Glocker et al., 2009; Lanternier et al., 2013). Finally, patients with autoimmune polyendocrinopathy type 1 syndrome (APS-1 also called APECED syndrome) caused by biallelic mutations of AIRE have high titers of neutralizing autoantibodies against IL-17A, IL-17F and/or IL-22 and suffer from CMC as the only infectious disease (Kisand et al., 2010; Puel et al., 2010). Collectively, these experiments of Nature suggest that CMC is caused by impaired IL-17 immunity, at least in the setting of these various conditions and, possibly, in other clinical settings (Puel et al., 2012).
Patients genetically prone to CMC but normally resistant to most other infections are considered to have CMC disease (CMCD) (Canales et al., 1969; Kirkpatrick et al., 1971; Wells, 1970; Wells et al., 1972). The phenotype is not strictly limited to CMC, as these patients often display other infections, such as staphylococcal cutaneous disease, and even autoimmune manifestations, such as thyroiditis (Atkinson et al., 2001; Liu et al., 2011b). Moreover, this condition is not benign, as the patients may develop mucocutaneous carcinomas and cerebral aneurysms (Leroy et al., 1989; Williamson, 1969). Complete AR IL-17RA deficiency and partial AD IL-17F deficiency were the first two genetic etiologies of CMCD to be discovered (Puel et al., 2011). Heterozygous gain-of-function (GOF) mutations of STAT1 were recently discovered (Liu et al., 2011b; van de Veerdonk et al., 2011). They were rapidly found in about half of CMCD patients (Hori et al., 2012; Liu et al., 2011b; Romberg et al., 2013; Sampaio et al., 2013; Smeekens et al., 2011; Takezaki et al., 2012; Uzel et al., 2013; van de Veerdonk et al., 2011; Wang et al., 2013b) (data not shown). These patients also display impaired IL-17 T-cell development, although the mechanisms involved in this impairment remain unclear. Together, these studies unambiguously indicate that IL-17A and IL-17F are essential for protective mucocutaneous immunity to C. albicans and, to a lesser extent, S. aureus. However, no genetic etiology has yet been identified for about half the known patients with CMCD.
We studied two siblings with CMCD suffering from oral thrush due to C. albicans and recurrent blepharitis due to S. aureus. Both patients also displayed transient atopic dermatitis in infancy. We deciphered the etiology of CMCD in these patients by a genome-wide (GW) approach based on linkage analysis and exome sequencing (Bogunovic et al., 2012; Boisson et al., 2012; Bolze et al., 2010; Byun et al., 2010). We identified a biallelic missense mutation (T536I) in the adaptor molecule ACT1 (TRAF3IP2). This missense mutation, located in the SEFIR, impaired the homotypic interaction of ACT1 with the IL-17 receptors abolishing the response to IL-17A and IL-17F in fibroblasts and to IL-17E in leukocytes. In contrast, the T536I allele did not affect the interaction with other proteins (e.g. HSP90, HSP70 or CD40), which is mediated by other domains. Fibroblastic cells from healthy individuals for the homozygous D10N missense polymorphism had a diminished but not abolished response to IL-17A. IL-17A and IL-17F depend on ACT1 to mediate protective mucocutaneous immunity to C. albicans and S. aureus and the other IL-17 cytokines seem to be redundant in host defense.
Results
Whole-exome sequencing in CMCD patients reveals a homozygous missense mutation of ACT1
We investigated a 30-year-old man (P1) born to consanguineous Algerian parents (Figure 1A). At the age of eight years, he developed macrocheilitis associated with progressive chronic macroglossia. He has since suffered from recurrent oral candidiasis affecting the mouth and tongue. His sister is 28 years old (P2) and has suffered, since early childhood, from recurrent episodes of bilateral blepharitis caused by S. aureus and folliculitis decalvans (Quinquaud’s folliculitis) caused by S. aureus. She has also had occasional episodes of oral thrush due to Candida albicans and of onycomycosis due to Candida parapsilosis. Both patients had infantile seborrheic dermatitis, which resolved with local treatment within 6 weeks. Immunological investigations were unremarkable (case report and Table S1) A GW linkage (GWL) analysis by homozygosity mapping in these two patients and their two healthy siblings was combined with whole-exome sequencing (WES) for P2. Fifty-six homozygous regions, encompassing 93 Mb, were common to the two affected siblings but were not found in the two unaffected siblings (Figure 1B). Only three homozygous non synonymous coding region variants (in C6ORF183, DOPEY2 and ACT1) found in P2 were not found in public databases or in our in-house database (more than 10,000 chromosomes), indicating that the frequency of homozygosity for these alleles is less than 10−8 (Table S2). The ACT1 mutation was chosen for further investigation as a potential disease-causing gene, due to its potential impact on IL-17 pathways (Chang et al., 2006; Chang et al., 2011; Qian et al., 2007). ACT-1 is recruited to IL-17RA, IL-17RB and IL-17RC and activates the NF-κB, MAPK, and C/EBP pathways, leading to the induction of target genes in keratinocytes, epithelial cells and fibroblasts stimulated with IL-17 cytokines (Gaffen, 2009; Maitra et al., 2007; Onishi and Gaffen, 2010). P1 and P2 were found to be homozygous for the c.1607C>T variant of ACT1, leading to production of the p. T536I protein (Figure 1C). The intrafamilial segregation pattern was consistent with an AR trait (Figure 1A–C). The threonine residue in position 536 has been conserved throughout evolution (Figure 1D). Moreover, two computer programs (Polyphen, SIFT) predicted the T536I mutation to be deleterious (Adzhubei et al., 2013; Ng and Henikoff, 2003). The p. T536I mutation affects the C-terminal part of the SEFIR domain, which is required for the recruitment of ACT1 to IL-17RA, IL-17RB, IL-17RC, IL-17RD and IL-17RE (Figure 1E) (Gaffen, 2009; Mellett et al., 2012).
Figure 1. A kindred with CMCD and autosomal recessive ACT1 deficiency.

(A) Pedigree of the kindred showing the segregation of the ACT1 missense allele T536I. Individuals for whom genetic status could not be evaluated are indicated by “?”. Generations are designated by Roman numerals and individuals by Arabic numerals. (B) Manhattan plot showing the results for linkage analyses. The Lodscore (log10) for each SNP is plotted against chromosomal position (x-axis) for this kindred. The three genes carrying an unreported missense mutation identified by WES in the positive regions are indicated. (see also Table S2) (C) ACT1 DNA sequence electropherograms, for a healthy control and the four members of the kindred, for the region corresponding to the missense mutation (D) Alignment of ACT1 orthologs from different species. (E) Schematic diagram of the ACT1 protein. The gray box indicates the TRAF-interacting domain. The helix-loop-helix (HLH), U3-box (U3-box) and similar expression to fibroblasts growth factor and IL-17R (SEFIR) domains are shown. The arrow indicates the position of the missense mutation.
The T536I ACT1 mutation impairs homotypic interactions with IL-17 receptors
We assessed the amount of human ACT1 (hACT1) protein in SV40-immortalized fibroblasts (SV-40 fibroblasts) and Epstein-Barr virus-immortalized B-cell lines (EBV-B cells) from four controls, P2 and a patient with complete IL-17RA deficiency (IL17RA−/−). All cells tested contained similar amounts of hACT1 protein (Figure 2A). The cellular localization of T536I-hACT1 in SV-40 fibroblasts from P2 is similar to that of WT hACT1 in control fibroblasts (Figure S1A). The interaction between IL-17 receptors and ACT1 is dependent on the homotypic dimerization of two SEFIR domains. Although T536 is not conserved in all protein paralogs harboring a SEFIR domain (Novatchkova et al., 2003), we hypothesized that the T536I missense mutation might impair the homotypic interaction of ACT1 with IL-17 receptors. HEK293T cells were cotransfected with WT-hACT1-HA or T536I-hACT1-HA and human IL-17RA-V5, IL-17RB-V5, or IL-17RC-V5 vectors. Immunoprecipitation with an anti-HA antibody showed that the T536I-hACT1-HA protein, unlike WT-hACT1-HA, was not bound to IL-17RA, IL-17RB or IL-17RC or was only weakly bound to these receptors (Figure 2B–2D). The SEFIR domain also plays an important role in ACT1 homodimerization (Liu et al., 2011a; Mauro et al., 2003). We thus assessed the capacity of the T536I protein to homodimerize in HEK293T cells, by cotransfecting these cells with WT-hACT1-HA or T536I-hACT1-HA and WT-hACT1-FLAG or T536I-hACT1-FLAG. Coimmunoprecipitation with the HA-antibody showed that hACT1 proteins with and without the T536I substitution were equally capable of homodimerization (Figure 2E, Figure S1B). ACT1 also binds to HSP90 via its N-terminus (Wang et al., 2013a). We assessed the impact of the T536I mutation on this interaction in HEK293T cells, by cotransfecting these cells with WT-hACT1-HA or T536I-hACT1-HA-tagged and hHSP90-FLAG. Co-immunoprecipitation with the anti-HA antibody showed that the interaction between ACT1 and HSP90 or HSP70 was intact (Figure 2F). Finally, T536I-hACT1 did not affect the stability of ACT1-interacting proteins such as TRAF2, TRAF3, TRAF5, TRAF6, SF2 or IL-17RA (Figure S1C and S1D). Thus, the T536I missense mutation selectively prevents the homotypic binding of ACT1 with at least three IL-17 receptor chains.
Figure 2. The T536I mutation impairs ACT1 recruitment to the IL-17 receptors.

(A) ACT1 expression in EBV-B cells and SV-40 fibroblasts from P2. HEK293T cells transfected with WT-hACT1 or empty vector are shown as a control for the specificity of anti-ACT1 antibody. GAPDH is used as a loading control. (B–D) Immunoprecipitation of HA-tagged hACT1 in Act1−/− cells reconstituted with WT-hACT1-HA or T536I-hACT1-HA. Cells are cotransfected with IL-17RA-, IL-17-RB- or IL-17RC-V5-tagged receptor chains. Immunoblotting analyses were performed with anti-HA or anti-V5 specific antibodies (n=3). (E) Homodimerization of ACT1. hACT1-HA was immunoprecipitated in HEK293T cells overproducing WT- or T536I-hACT1-HA and WT- or T536I-hACT1-FLAG. The immunoblotting analysis was performed with specific anti-HA, anti-FLAG or anti-HSP90 antibodies (n=2). (F) Heterodimerization of hACT1 and HSP90. WT- or T536I-hACT1-HA was immunoprecipitated in HEK293T cells overproducing WT- or T536I-hACT1-HA and HSP90-FLAG. Immunoblotting analyses were performed with specific anti-HA, anti-FLAG or anti-HSP70 antibodies (n=2). (see also Figure S1).
The missense T536I mutation of ACT1 impairs the fibroblast response to IL-17
ACT1 is an essential component of the IL-17A response pathway and is required for the secretion of IL-6 and GRO-α in mice, via activation of the IKK and MAPK pathways (Chang et al., 2006). We first assessed the activation of the IL-17A response pathway in Act1−/− cells transfected with mock, WT-mAct1 or T517I-mAct1 plasmids. Likewise, the expression of the orthologous missense mouse Act1 (mACT1) allele (T517I) in Act1−/− was normal (Figure S2A). After stimulation with IL-17A, only WT-mACT1 restored activation of the IKK (IκBα, NF-κB-P65) and MAPK (JNK, ERK, P38) pathways (Figure S2B). We also assessed the effects of T536I-hACT1, by investigating the IL-17 pathways in SV40-fibroblasts from controls, P2 and an IL-17RA-deficient patient (Puel et al., 2011). SV40-fibroblasts from P2, like IL-17RA-deficient fibroblasts, produced no IL-6 or GRO-α (also known as KC or CXCL1) in response to various doses of IL-17A (Figure 3A) or IL-17F (Figure S2C), whereas control fibroblasts responded to even the lowest doses (10 ng/ml). Cells from P2 and the IL-17RA-deficient patient responded normally to IL-1β. In a similar experiment in Act1−/− cells, WT-mACT1 induced the expression of Cxcl1 (encoding GRO-α), I andl6 Csf2 (encoding GM-CSF), whereas T517I-mACT1 did not (Figure S2D). We also assessed the activation of the NF-κB pathways upon transfection of an NF-κB driven luciferase construct in SV-40 fibroblasts. Upon IL-17A, we found no luciferase activity in hACT1-T536I-mutated or IL-17RA-deficient fibroblasts whereas controls cells responded (Figure S2E). We then transduced SV40-fibroblasts from P2 with retroviruses expressing WT-hACT1. The stable production of WT-hACT1 in the cells of P2 restored the induction of IL-6 and GRO-α production in response to IL-17A to amounts similar to those observed in the control (Figure 3B). No such increase in the production of these molecules was observed after transduction with a mock retrovirus. By contrast, the introduction of the WT-hACT1 allele did not lead to the production of IL-6 and GRO-α in IL-17RA-deficient cells (Figure 3B). ACT1 also acts in complex with SF2, to prolong the half-life of mRNAs upon costimulation with IL-17A and TNF-α, sustaining the production of cytokines, such as GRO-α and IL-6 (Sun et al., 2011). We thus investigated indirectly this synergistic effect by measuring the production of GRO-α and IL-6 in response to costimulation with IL-17A and TNF-α. We used hACT1-mutated fibroblasts from P2 transduced with an empty or WT-hACT1-expressing virus. The P2 fibroblasts re-expressing WT-hACT1 displayed synergic effects of IL-17A and TNF-α, secreting more GRO-α and IL-6 than in response to either cytokine alone (Figure 3B). This synergic effect was not detectable in cells from P2 transduced with an empty vector or in IL-17RA-deficient cells transduced with an empty or WT-hACT1-encoding vector (Figure 3B). In Act1−/− cells, the T517I-mACT1 did not prolong the half-life of Gro-α and I mRNAl6 after treatment with IL-17A and TNF-α (Figure S2E). Collectively, there is therefore a defect of transcriptional up-regulation of the IL-17 target genes, due to impaired NF-κB activation in the fibroblasts of the CMCD siblings, homozygous for the T536I mutant allele of ACT1.
Figure 3. The T536I ACT1 mutation abolishes the IL-17-responsive pathway in fibroblasts.
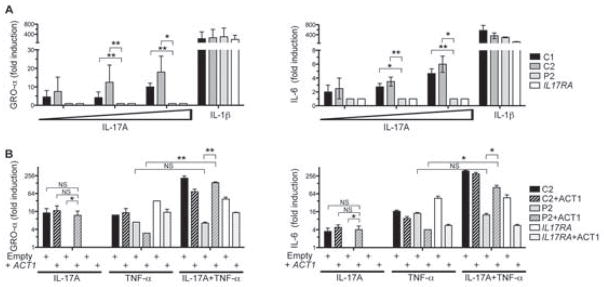
(A) IL-6 and GRO-α quantification in the supernatant of SV-40 fibroblasts from controls (C1, C2), P2 or IL-17RA-deficient patients, in response to IL-17A (10, 100 and 1,000 ng/ml) or IL-1β (10 ng/ml); errors bars, SEM (n=3). (B) Complementation of the fibroblasts of P2 with the WT ACT1 allele. IL-6 and GRO-α quantification in the supernatant of SV-40 fibroblasts from the control, P2 or IL-17RA-deficient patients complemented (+ACT1) or not complemented with the WT ACT1 allele, after stimulation with IL-17A (100 ng/ml), TNF-α (2 ng/ml) or both. Errors bars, SEM (n=3). Statistical analyses were performed by the unpaired t-test method (NS: not significant; *, P<0.05; **, P< 0.01). (see also Figure S2)
The missense D10N polymorphic allele of ACT1 is hypomorphic, but not null
Recent studies have investigated the polymorphic D10N allele of human ACT1 (rs33980500), which is associated with an increased risk of psoriasis and psoriatic arthritis (Ellinghaus et al., 2010; Huffmeier et al., 2010). Studies based on overexpression in HEK293T cells or in Act1−/− cells suggest that this allele is loss-of-function, at least for the interaction with TRAF-6 and the activation of the IL-17A-dependent gene expression (Huffmeier et al., 2010; Wang et al., 2013a). Due to the frequency of the D10N allele in most human populations studied (MAF about 0.09), about 0.8% of the population worldwide is predicted to be homozygous. If this were a null mutation, abolishing cellular responses to IL-17 in particular, the high frequency of homozygotes would not be consistent with the rarity of CMCD (about 1/100,000). We investigated this apparent discordance, by testing an individual without CMCD carrying two copies of the polymorphic D10N allele. We evaluated the response of SV40-fibroblasts from this individual to IL-17A. The cells from the individual with the D10N allele responded weakly to IL-17A, with low but detectable GRO-α and IL-6 induction (Figure 4A–B). Moreover, costimulation with IL-17A and TNF-α was synergistic in the cells of the individual with the D10N allele, contrasting with the complete absence of a synergistic response in cells from P2 and the IL-17RA-deficient patient. These findings demonstrate that the human D10N ACT1 allele is hypomorphic, but not null, for cellular responses to IL-17A, alone or in combination with TNF-α. This result reconciles the frequency of homozygosity for the D10N allele in the human population and the rarity of CMCD. It also highlights the uniqueness of the T536I allele, which is a true null allele for IL-17 responses.
Figure 4. The D10N ACT1 polymorphism impairs but do not abolishes he IL-17-responsive pathway in fibroblasts.
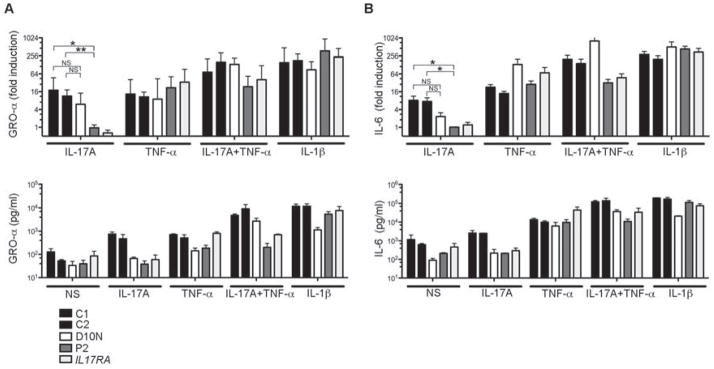
(A) GRO-α and (B) IL-6 quantification in the supernatant of SV-40 fibroblasts from controls, D10N individual, P2 and an IL-17RA-deficient patient, upon stimulation with IL-17A (500 ng/ml), TNF-α (2 ng/ml) or both (IL-17A+TNF-α), or IL-1β (10 ng/ml); errors bars, SEM (n=7). Statistical analyses were performed by the unpaired t-test method (NS: not significant; *, P<0.05; **, P< 0.01).
The T536I missense mutation of ACT1 impairs the IL-17 signaling pathway in leukocytes
Some IL-17 cytokines also act on leukocytes and contribute to the differentiation of certain cytokine-producing T cells. IL-17E (also known as IL-25) has recently been shown to signal through IL-17RA and IL-17RB, leading to the differentiation of CD4+ T cells into IL-5- and IL13-producing cells in mice (Rickel et al., 2008). We investigated the IL-17E response pathway in PBMCs from controls, P1, P2 and healthy members of their family. Thymic stromal lymphopoietin (TSLP)-stimulated PBMCs have been shown to produce IL-5 and IL-13 in presence of IL-2 or IL-17E (Rickel et al., 2008). Control PBMCs produced about five times more IL-5 in response to costimulation with IL-2 and IL-17E than in response to each cytokine individually (Figure 5A). By contrast, cells from P1 and P2 were unable to respond synergistically to the combination of IL-2 and IL-17E, whereas IL-2 alone induced IL-5 production. The patients’ healthy sisters (II.2 and II.3) responded normally to IL-17E, whereas the IL17RA-deficient patient did not (Figure 5A). Thus, the T536I missense mutation of ACT1 also impaired IL-17 responses in T cells. Act1−/− mice have higher proportions of IL-17A-, IL-17F-, IL-22-, and IFN-γ-producing T cells than control mice, but the underlying mechanisms are unknown (Qian et al., 2007; Wang et al., 2013a). We therefore carried out flow cytometry to investigate, ex vivo and in vitro, the proportion of IL-17A-, IL-22- and IFN-γ-producing CD3+ T cells. P1 had higher proportions of IL-17A-producing (9.6%) and IL-22-producing (7.8%) T cells ex vivo than all 85 healthy controls tested (Figure 5B), which is consistent with findings for the mouse model (Qian et al., 2007). By contrast, this patient had normal proportions of IFN-γ–producing T cells. Moreover, T cells from P1 and P2 cultured with IL-2, IL-17E, or both, and restimulated by incubation with PMA/ionomycin for 12 h, also contained higher proportions of IL-17A-producing T cells than T cells from the controls, the two healthy siblings, and the IL17-RA-deficient patient (Figure S3). By contrast, the proportions of IFN-γ- and IL-13-producing T cells were normal (Figure S3). Despite the large proportion of IL-17-producing T cells in the two siblings with ACT1 mutations and CMCD, these individuals had no IL-17 response and were not protected against C. albicans. The expansion of the IL-22-producing T-cell population does not seem to be sufficient to control CMC and seems to have no other detectable clinical consequences.
Figure 5. The T536I ACT1 mutation abolishes the IL-17E response in T cells.

(A) PBMCs from controls, patients (P1, P2) and healthy relatives (II.2, II.3) and from an IL-17RA-deficient patient were cultured in TSLP for 24 h, harvested, and restimulated with IL-2 and IL-17E for an additional 72 hours. IL-5 concentrations in the culture supernatants were determined by ELISA. IL-5 concentrations were normalized to 100 pg/ml upon IL-2 stimulation. Errors bars, SEM (n=2). (B) Percentages of IL-17A+-, IL-22+- and IFN-γ+- producing CD3+ cells, as determined by flow cytometry, in nonadherent PBMCs activated by incubation for 12 h with PMA and ionomycin. Each symbol represents a value from a healthy control individual (black circles), P1 (black rectangle) or an IL-17RA-deficient patient (black upside-down triangles). (see also Figure S3).
The T536I missense mutation of ACT1 does not impair the CD40 response pathway
ACT1 has also been reported to regulate the CD40 and BAFF-R pathways (Qian et al., 2004). Act1−/− mice (BALB/c/129 background) display enhanced B cell responses to CD40L and BAFF. As mACT1 has been shown to be recruited to TRAF3 via its TRAF-interacting domain (Qian et al., 2004), it has also been suggested that ACT1 inhibits CD40 and BAFF-R through interaction with TRAF3 (Qian et al., 2004). We thus assessed the impact of the T536I mutation on this interaction in HEK293T cells cotransfected with hCD40-FLAG and hACT1-HA with and without the T536I mutation. The T536I mutation did not abolish the interaction of ACT1 with the costimulatory molecule CD40 (Figure 6A). Moreover, the time course of IKK and IκBα phosphorylation upon CD40L stimulation was similar in EBV-B cells from controls (C1 and C2) and from P2 (Figure 6B). These data, obtained with EBV-B cells in vitro, are consistent with the lack of an overt B cell phenotype in vivo. Indeed, Act1−/− mice have hypergammaglobulinemia, with high serum concentrations of IgG, IgA and IgE. Similarly, serum IgG concentrations were high in P1 and P2 (>17 g/l), but similar to those in patients with chronic infections. By contrast, serum IgE concentrations were normal in P1 and P2 (< 150 kIU/L; Table S1). Likewise, another independent Act1−/− mouse line in another genetic background (129/Sv) presented no overt B cell phenotype (Claudio et al., 2009). Overall, the missense T536 mutation in ACT1 did not significantly enhance the CD40 and BAFF-R pathways in the patients’ B cells. Thus this mutation, affecting the SEFIR domain, selectively impairs the IL-17 response pathway in leukocytes, such as T cells, and in non-hematopoietic cells, such as fibroblasts.
Figure 6. The T536I-ACT1 mutation does not perturb the CD40-responsive pathway.

(A) Immunoprecipitation of ACT1-HA in HEK293T cells reconstituted with WT-hACT1-HA or T536I-hACT1-HA. Cells were cotransfected with human CD40-FLAG. Immunoblotting analyses were performed with anti-HA, anti-TRAF3 or anti-FLAG specific antibodies. (B) Activation of CD40 pathways in EBV-B cells from P2. Immunoblotting analysis was performed using antibodies against phospho-IKKα and phospho-IKKβ (P-IKKs), IκBα and phospho-p42 and phospho-p44 (P-p42-p44) in CD40L-stimulated EBV-B cells from the control or P2 (n=2).
Discussion
We identified biallelic mutations of ACT1 as a new genetic etiology of CMCD in two patients. The morbid T536I mutation disrupts the interaction of ACT1 with IL-17 receptors, but not with CD40 and BAFF-R. This mutation affects the SEFIR domain, which has been characterized and modeled with computer software (Liu et al., 2011a; Zhang et al., 2013). Deletion of the terminal part of the mACT1 SEFIR domain, containing the amino acid corresponding to the T536 residue of the human protein, did not affect the interaction of ACT1 with IL-17 receptors (Liu et al., 2011a). However, introduction of the orthologous missense mutation into mAct1 (T517I) abolished the induction of cytokines in response to IL-17A stimulation (this report). These data therefore suggest that the human T536 and mouse T517 residues are essential for the conformational structure of the SEFIR domain, or that their replacement by an isoleucine residue disrupts the activity of this domain. The effects of this mutation also strongly suggest that ACT1 homodimerization and ACT1-IL-17R interaction involve different regions of the SEFIR domain. Indeed, the T536I mutation leads to a loss of homotypic interaction with members of the IL-17 receptor family but does not impair homodimerization.
This observation explains the patients’ CMCD phenotype. Indeed, patients with AR IL-17RA or AD IL-17F deficiencies suffer from CMCD due to impaired IL-17A- and IL-17F-mediated immunity (Puel et al., 2011) and patients heterozygous for GOF STAT1 alleles also display CMCD due to impaired IL-17 immunity (Liu et al., 2011b; Okada et al., 2013; Romberg et al., 2013; Smeekens et al., 2011; Takezaki et al., 2012; van de Veerdonk et al., 2011). Biallelic mutations of ACT1 therefore define the fourth genetic etiology of CMCD and the second affecting the IL-17 response pathway. In both mice (Qian et al., 2007) and humans (this report), ACT1 deficiency leads to expansion of IL-17- and IL-22-producing T cell populations. The mechanism underlying this phenotype is unknown. High amounts of IL-17 are ineffective in the absence of ACT1, and IL-22, the function of which is unknown in humans, does not seem to compensate for the lack of IL-17 responses. The clinical phenotypes of IL-17RA and ACT1 deficiencies are indistinguishable, at least in terms of the CMC and staphylococcal disease of the patients.
The ACT1-T536I-mutated patients also developed mild and transient infantile seborrheic dermatitis, a relatively common skin disease. Among the possible causes of infantile seborrheic dermatitis, infection by Malassezia furfur and Staphylococci have both been suspected to be causal (Bieber, 2008). This condition is rare in developed countries, thanks to the improvement of hygiene (Kalliomaki et al., 2001). The mild infantile dermatitis seen in the ACT1-T536I-mutated patients might perhaps also result, at least in part, from the expansion of the IL-22 T cell population, as suggested by findings in humans (Nograles et al., 2009) and mice (Wang et al., 2013a). The lack of infantile dermatitis in IL-17RA-deficient patients, whose IL-22-producing T cells are not expanded, also favors this hypothesis (Puel et al., 2011). Too few IL-17RA-deficient patients have been diagnosed yet to draw firm conclusions, however. Two strains of Act1−/− mice were found to have atopic dermatitis (Matsushima et al., 2010; Qian et al., 2008; Qian et al., 2004; Wang et al., 2013a); this was not observed in a third strain (Claudio et al., 2009). The ACT1-T536I-mutated patients did not display any atopy.
We also found that the D10N variant of ACT1 was hypomorphic, but not null, for cellular responses to IL-17, consistent with its high frequency in human populations and the rarity of CMCD. There is an apparent discrepancy between our data and others (Ellinghaus et al., 2010; Huffmeier et al., 2010; Wang et al., 2013a), which proposed that this psoriasis susceptibility allele is functionally null. This discrepancy might be explained by the presence of two ACT1 isoforms in humans, a short form and a long form, which contains nine additional amino acids at the N-terminus; there is only one short isoform of Act1 in mouse. Only the shortest D10N isoform has been demonstrated to be null. The longest (D19N) might be functional. Further studies are required to clarify this matter. The two patients homozygous for the T536I allele of ACT1 do not display psoriasis suggesting the absence of IL-17 activity is not the factor triggering psoriasis. In any case, ACT1 is required for IL-17A and IL-17F mucocutaneous immunity to C. albicans and S. aureus in humans.
Other IL-17 cytokines have been implicated in immunity. IL-17B, IL-17C and IL-17E have been shown to bind to IL-17R family members (IL-17RA, IL-17RB, IL-17RE) requiring ACT1, for signaling in mice (Gaffen, 2009). No data are available for IL-17D. The cells of patients with ACT1 mutations are probably unable to respond to IL-17C, as the mutated ACT1 does not bind to IL-17RA (Chang et al., 2011). The cells of our patients failed to respond to IL-17E, and the response to this cytokine is IL-17RB-dependent in mice (Rickel et al., 2008). These cells are probably also unable to respond to IL-17B, which also binds IL-17RB in human cells (Shi et al., 2000). Collectively, the observation that IL-17RA-deficient and ACT1-mutated patients display CMCD as their only major infection suggests that IL-17B, C, D, and E play largely redundant roles in host defense in humans. This is intriguing, as IL-17C and IL-17E have been implicated in immunity to worms and bacteria in mice. For example, Il17re−/− mice (unresponsive to IL-17C) die prematurely after Citrobacter rodentium infection, highlighting the role of IL-17C in intestinal immunity (Chang et al., 2011; Ramirez-Carrozzi et al., 2011; Song et al., 2011). Moreover, Il25−/− mice (lacking IL-17E) are more susceptible to Nippostrongylus brasiliensis and Trichuris muris than control mice (Fallon et al., 2006; Owyang et al., 2006; Zhao et al., 2010). Act1−/− and conditional K18creAct1−/− mice (in which ACT1 is deleted in epithelial cells) are also more susceptible to infection with Nippostrongylus brasiliensis (Kang et al., 2012). ACT1 plays a broader role than IL-17RA in IL-17 responses, but patients with ACT1 mutations do not suffer from a broader range of infections than patients with IL-17RA and IL-17F deficiencies (with impaired IL-17A and IL-17F immunity) (Puel et al., 2011). We need to diagnose and to study a larger number of patients before we can draw any firm conclusions, but our observations suggest that IL-17B, C, D and E are largely redundant. An alternative, intriguing possibility is that these human cytokines can also signal via ACT1-independent pathways.
Methods
Sample collection
This study was conducted in accordance with the Helsinki Declaration, with informed consent obtained from each patient or the patient’s family. The study was approved by the local ethics committee of Necker-Enfants Malades Hospital, Paris, France and the Rockefeller University Hospital, New York, USA.
Genetic analysis
Genotyping and linkage analysis
Four members of this Algerian family were genotyped with the Affymetrix Genome-wide SNP 6.0 array. Genotype calling was achieved with Affymetrix Power Tools (http://www.affymetrix.com/partners_programs/programs/developer/tools/powertools.affx) for the four family members. We discarded monomorphic SNPs, SNPs with a call rate lower than 100% and SNPs presenting more than one Mendelian inconsistency in the family. SNPs were further filtered with population-based filters (Purcell et al., 2007). We then used about 110,347 high-quality SNP markers to carry out linkage analysis, assuming autosomal recessive inheritance with complete penetrance (homozygosity mapping). Parametric multipoint linkage analysis was carried out with the Merlin program (Abecasis et al., 2002). The Algerian family founders and HapMap CEU trios were used to estimate allele frequencies and to define linkage clusters, with an r2 threshold of 0.4.
Massively parallel sequencing
Genomic DNA (3 μg) extracted from the peripheral blood cells of the patient was sheared with a Covaris S2 Ultrasonicator (Covaris). An adapter-ligated library was prepared with the Paired-End Sample Prep kit V1 (Illumina). Exome capture was performed with the SureSelect Human All Exon kit (Agilent Technologies). Single-end sequencing was performed on an Illumina Genome Analyzer IIx (Illumina), generating 72-base reads.
Sequence alignment, variant calling and annotation
The sequences were aligned with the human genome reference sequence (hg19 build), using BWA aligner (Li and Durbin, 2009). Downstream processing was carried out with the Genome analysis toolkit (GATK) (McKenna et al., 2010) SAMtools (Li et al., 2009) and Picard Tools (http://picard.sourceforge.net). Substitution calls were made with a GATK UnifiedGenotyper, whereas indel calls were made with a GATK IndelGenotyperV2. All calls with a read coverage ≤2x and a Phred-scaled SNP quality of ≤20 were filtered out. All the variants were annotated with the GATK Genomic Annotator.
Cell lines, immortalization and complementation
Control and patient-derived fibroblasts were immortalized by transfection with a plasmid containing the simian virus 40 large T antigen gene. Transformed cell lines were grown in DMEM (GIBCO®#10566) supplemented with 10% fetal calf serum (GIBCO®#16000). The IL17RA−/− fibroblasts used have been described elsewhere (Puel et al., 2011). ACT1 mRNA was reverse transcribed, amplified and inserted into the retroviral vector pMSCV (Clontech). Infectious viral particles were produced by cotransfecting GP2-293 packaging cells with pVSV-G and the retroviral expression vector pMSCV-hACT1 or an empty vector (Clontech). Viral particles were collected between 48 and 72 hours after transfection and were used to infect SV40-transformed fibroblasts. Infected cells were selected on medium supplemented with 0.4–0.8 μg/ml puromycin.
Cell lysis, immunoprecipitation and immunoblotting
Cells were lysed in a lysis buffer containing 30 mM Tris-HCl pH 7.5, 120 mM NaCl, 2 mM KCl, 1% Triton X-100 and 2 mM EDTA supplemented with protease inhibitor cocktail (Complete, Roche) and phosphatase inhibitor cocktail (PhoStop, Roche). Laemmli buffer supplemented with DTT was added to clear supernatants. For immunoprecipitation, cells were lysed in lysis buffer [0.5% Triton X-100, 20 mM Hepes (pH 7.4), 150 mM NaCl, 12.5 mM β-glycerophosphate, 1.5 mM MgCl2, 10 mM NaF, 2 mM dithiothreitol, 1 mM sodium orthovanadate, 2 mM EGTA, 20 mM aprotinin, 1 mM phenylmethylsulfonyl fluoride]. Cell extracts were incubated with 1 μg of the appropriate antibodies overnight at 4°C with 20 ul of protein A Sepharose beads. After incubation, beads were washed four times with lysis buffer. Samples were separated by SDS–PAGE and proteins were transferred onto a PVDF membrane. The membrane was blocked in TBS supplemented with 0.1% Tween 20 and 5% skimmed milk powder and incubated with the primary antibody, followed by the appropriate horseradish peroxidase-conjugated secondary antibody. Immunoreactive proteins were visualized by enhanced chemiluminescence.
Luciferase assays
For NF-κB-dependent reporter assays, SV40-transformed fibroblasts were transiently transfected with the NF-κB-dependent firefly luciferase plasmid pGL4.32 (Promega) and the Renilla luciferase plasmid as internal control using Lipofectamine™ LTX reagent (Invitrogen). After 24 hours, cells were stimulated with TNF or IL-1β for four hours and luciferase activities were assessed using the dual luciferase assay kit (Promega).
PBMC culture
Frozen PBMCs were cultured in the presence of 100 ng/ml thymic stromal lymphopoietin (Amgen) in X-VIVO 15 (Lonza) plus 5% human AB serum (Lonza), as previously described (Rickel et al., 2008). PBMCs were collected, washed and resuspended at a density of 4 × 106 cells/well in 48-well plates with a final volume of 0.5 ml/well, in the presence of 10 ng/ml recombinant human IL-2 (R&D Systems) and 10 ng/ml recombinant human IL-17E (R&D Systems). After three days, the amount of IL-5 secretion was determined with ELISA kits (DY205, R&D Systems).
Flow cytometry
PBMCs cultured as described above were then stimulated with PMA+ionomycin (10−7 M/10−5 M/100 μg/ml) or left unstimulated. Concomitantly to stimulation, protein transport was blocked with GolgiStop and GolgiPLug (BD). Cells were stained with anti-CD3 (BD, #560366) and anti-CD19 (BD, #340951) antibodies for cellular phenotyping, and with Aqua Live/Dead (Invitrogen) to exclude dead cells. The cells were fixed and permeabilized (BD, #554722), and then stained with antibodies against IL-17A (ebioscience, #53-7179-42), IL-13 (Biolegends, #501907) and IFN-γ (BD, #559326). Stained PBMCs were captured by flow cytometry with a BD LSRII flow cytometer and FACS Diva software. The data were analyzed with FlowJo (©Tree Star).
Statistical analysis
Data were analyzed with Student’s t test using GraphPad Prism (GraphPad Software) and p < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank the patients and their family for participating in the study, all the member of both branches of the laboratory for discussions, especially, Maya Chrabieh, Malik Bensifi, Marcela Moncada Velez, Tatiana Kochetkov, Minji Byun, Fanny Lanternier, Stephanie Boisson-Dupuis, Dusan Bogunovic and Alexandre Bolze. We thank Yelena Nemiroskaya, Eric Anderson and Tiffany Nivare for their assistance. We also thank the Alison North and Kaye Thomas from the Bio-Imaging Resource Center at the Rockefeller University. The Laboratory of Human Genetics of Infectious Diseases is supported by grants from the European Research Council (ERC-2010-AdG-268777), Institut National de la Santé et de la Recherche Médicale, University Paris Descartes, the St. Giles Foundation, the National Center for Research Resources and the National Center for Advancing Sciences (NCATS) grant number 8UL1TR000043 from the National Institutes of Health, the Jeffrey Modell Foundation and the Rockefeller University. S.C. was supported by the AXA Research Fund.
Footnotes
Contributions
B.B., C.W., L.W. and A.P. performed experiments. V.P. performed the GWL analysis. C.F., C.P. and M.R provided all the clinical data for the patients. S.C., C.P., A.B., A.P., L.A. and X.L. provided reagents and suggestions. C.F. and C.P. performed immunological explorations. B.B., L.A., X.L. and J.-L.C. coordinated the study, and B.B. and J.-L.C. wrote the manuscript. All authors discussed the results and commented on the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin--rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- Adzhubei I, Jordan DM, Sunyaev SR. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr Protoc Hum Genet. 2013;Chapter 7(Unit 7):20. doi: 10.1002/0471142905.hg0720s76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson TP, Schaffer AA, Grimbacher B, Schroeder HW, Jr, Woellner C, Zerbe CS, Puck JM. An immune defect causing dominant chronic mucocutaneous candidiasis and thyroid disease maps to chromosome 2p in a single family. Am J Hum Genet. 2001;69:791–803. doi: 10.1086/323611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieber T. Atopic dermatitis. N Engl J Med. 2008;358:1483–1494. doi: 10.1056/NEJMra074081. [DOI] [PubMed] [Google Scholar]
- Bogunovic D, Byun M, Durfee LA, Abhyankar A, Sanal O, Mansouri D, Salem S, Radovanovic I, Grant AV, Adimi P, et al. Mycobacterial Disease and Impaired IFN-gamma Immunity in Humans with Inherited ISG15 Deficiency. Science. 2012 doi: 10.1126/science.1224026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisson B, Laplantine E, Prando C, Giliani S, Israelsson E, Xu Z, Abhyankar A, Israel L, Trevejo-Nunez G, Bogunovic D, et al. Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL-1 and LUBAC deficiency. Nat Immunol. 2012;13:1178–1186. doi: 10.1038/ni.2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolze A, Byun M, McDonald D, Morgan NV, Abhyankar A, Premkumar L, Puel A, Bacon CM, Rieux-Laucat F, Pang K, et al. Whole-exome-sequencing-based discovery of human FADD deficiency. Am J Hum Genet. 2010;87:873–881. doi: 10.1016/j.ajhg.2010.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byun M, Abhyankar A, Lelarge V, Plancoulaine S, Palanduz A, Telhan L, Boisson B, Picard C, Dewell S, Zhao C, et al. Whole-exome sequencing-based discovery of STIM1 deficiency in a child with fatal classic Kaposi sarcoma. J Exp Med. 2010;207:2307–2312. doi: 10.1084/jem.20101597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canales L, Middlemas RO, 3rd, Louro JM, South MA. Immunological observations in chronic mucocutaneous candidiasis. Lancet. 1969;2:567–571. doi: 10.1016/s0140-6736(69)90264-5. [DOI] [PubMed] [Google Scholar]
- Chandesris MO, Melki I, Natividad A, Puel A, Fieschi C, Yun L, Thumerelle C, Oksenhendler E, Boutboul D, Thomas C, et al. Autosomal Dominant STAT3 Deficiency and Hyper-IgE Syndrome: Molecular, Cellular, and Clinical Features From a French National Survey. Medicine (Baltimore) 2012;91:e1–e19. doi: 10.1097/MD.0b013e31825f95b9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang SH, Park H, Dong C. Act1 adaptor protein is an immediate and essential signaling component of interleukin-17 receptor. J Biol Chem. 2006;281:35603–35607. doi: 10.1074/jbc.C600256200. [DOI] [PubMed] [Google Scholar]
- Chang SH, Reynolds JM, Pappu BP, Chen G, Martinez GJ, Dong C. Interleukin-17C promotes Th17 cell responses and autoimmune disease via interleukin-17 receptor E. Immunity. 2011;35:611–621. doi: 10.1016/j.immuni.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claudio E, Sonder SU, Saret S, Carvalho G, Ramalingam TR, Wynn TA, Chariot A, Garcia-Perganeda A, Leonardi A, Paun A, et al. The adaptor protein CIKS/Act1 is essential for IL-25-mediated allergic airway inflammation. J Immunol. 2009;182:1617–1630. doi: 10.4049/jimmunol.182.3.1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Beaucoudrey L, Puel A, Filipe-Santos O, Cobat A, Ghandil P, Chrabieh M, Feinberg J, von Bernuth H, Samarina A, Janniere L, et al. Mutations in STAT3 and IL12RB1 impair the development of human IL-17-producing T cells. J Exp Med. 2008;205:1543–1550. doi: 10.1084/jem.20080321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Beaucoudrey L, Samarina A, Bustamante J, Cobat A, Boisson-Dupuis S, Feinberg J, Al-Muhsen S, Janniere L, Rose Y, de Suremain M, et al. Revisiting human IL-12Rbeta1 deficiency: a survey of 141 patients from 30 countries. Medicine (Baltimore) 2010;89:381–402. doi: 10.1097/MD.0b013e3181fdd832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drewniak A, Gazendam RP, Tool AT, van Houdt M, Jansen MH, van Hamme JL, van Leeuwen EM, Roos D, Scalais E, de Beaufort C, et al. Invasive fungal infection and impaired neutrophil killing in human CARD9 deficiency. Blood. 2013;121:2385–2392. doi: 10.1182/blood-2012-08-450551. [DOI] [PubMed] [Google Scholar]
- Ellinghaus E, Ellinghaus D, Stuart PE, Nair RP, Debrus S, Raelson JV, Belouchi M, Fournier H, Reinhard C, Ding J, et al. Genome-wide association study identifies a psoriasis susceptibility locus at TRAF3IP2. Nat Genet. 2010;42:991–995. doi: 10.1038/ng.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallon PG, Ballantyne SJ, Mangan NE, Barlow JL, Dasvarma A, Hewett DR, McIlgorm A, Jolin HE, McKenzie AN. Identification of an interleukin (IL)-25-dependent cell population that provides IL-4, IL-5, and IL-13 at the onset of helminth expulsion. J Exp Med. 2006;203:1105–1116. doi: 10.1084/jem.20051615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffen SL. Structure and signalling in the IL-17 receptor family. Nature Reviews Immunology. 2009;9:556–567. doi: 10.1038/nri2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glocker E, Grimbacher B. Chronic mucocutaneous candidiasis and congenital susceptibility to Candida. Curr Opin Allergy Clin Immunol. 2010;10:542–550. doi: 10.1097/ACI.0b013e32833fd74f. [DOI] [PubMed] [Google Scholar]
- Glocker EO, Hennigs A, Nabavi M, Schaffer AA, Woellner C, Salzer U, Pfeifer D, Veelken H, Warnatz K, Tahami F, et al. A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N Engl J Med. 2009;361:1727–1735. doi: 10.1056/NEJMoa0810719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori T, Ohnishi H, Teramoto T, Tsubouchi K, Naiki T, Hirose Y, Ohara O, Seishima M, Kaneko H, Fukao T, Kondo N. Autosomal-Dominant Chronic Mucocutaneous Candidiasis with STAT1-Mutation can be Complicated with Chronic Active Hepatitis and Hypothyroidism. J Clin Immunol. 2012 doi: 10.1007/s10875-012-9744-6. [DOI] [PubMed] [Google Scholar]
- Huffmeier U, Uebe S, Ekici AB, Bowes J, Giardina E, Korendowych E, Juneblad K, Apel M, McManus R, Ho P, et al. Common variants at TRAF3IP2 are associated with susceptibility to psoriatic arthritis and psoriasis. Nat Genet. 2010;42:996–999. doi: 10.1038/ng.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalliomaki M, Salminen S, Arvilommi H, Kero P, Koskinen P, Isolauri E. Probiotics in primary prevention of atopic disease: a randomised placebo-controlled trial. Lancet. 2001;357:1076–1079. doi: 10.1016/S0140-6736(00)04259-8. [DOI] [PubMed] [Google Scholar]
- Kang Z, Swaidani S, Yin W, Wang C, Barlow JL, Gulen MF, Bulek K, Do JS, Aronica M, McKenzie AN, et al. Epithelial cell-specific Act1 adaptor mediates interleukin-25-dependent helminth expulsion through expansion of Lin(−)c-Kit(+) innate cell population. Immunity. 2012;36:821–833. doi: 10.1016/j.immuni.2012.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick CH, Rich RR, Graw RG, Jr, Smith TK, Mickenberg I, Rogentine GN. Treatment of chronic mucocutaneous moniliasis by immunologic reconstitution. Clin Exp Immunol. 1971;9:733–748. [PMC free article] [PubMed] [Google Scholar]
- Kisand K, Boe Wolff AS, Podkrajsek KT, Tserel L, Link M, Kisand KV, Ersvaer E, Perheentupa J, Erichsen MM, Bratanic N, et al. Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. J Exp Med. 2010;207:299–308. doi: 10.1084/jem.20091669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanternier F, Pathan S, Vincent Q, Liu L, Cypowyj S, Prando C, Migaud M, Taibi L, Ammar-Khodja A, Boudghene Stambouli O, et al. Human deep dermatophytosis and inherited CARD9 deficiency. New England Journal of Medecine. 2013 doi: 10.1056/NEJMoa1208487. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy D, Dompmartin A, Houtteville JP, Theron J. Aneurysm associated with chronic mucocutaneous candidiasis during long-term therapy with ketoconazole. Dermatologica. 1989;178:43–46. doi: 10.1159/000248386. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilic D. Unravelling fungal immunity through primary immune deficiencies. Curr Opin Microbiol. 2012;15:420–426. doi: 10.1016/j.mib.2012.06.003. [DOI] [PubMed] [Google Scholar]
- Liu C, Swaidani S, Qian W, Kang Z, Sun P, Han Y, Wang C, Gulen MF, Yin W, Zhang C, et al. A CC’ loop decoy peptide blocks the interaction between Act1 and IL-17RA to attenuate IL-17- and IL-25-induced inflammation. Sci Signal. 2011a;4:ra72. doi: 10.1126/scisignal.2001843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Okada S, Kong XF, Kreins AY, Cypowyj S, Abhyankar A, Toubiana J, Itan Y, Audry M, Nitschke P, et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med. 2011b;208:1635–1648. doi: 10.1084/jem.20110958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma CS, Chew GY, Simpson N, Priyadarshi A, Wong M, Grimbacher B, Fulcher DA, Tangye SG, Cook MC. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J Exp Med. 2008;205:1551–1557. doi: 10.1084/jem.20080218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maitra A, Shen F, Hanel W, Mossman K, Tocker J, Swart D, Gaffen SL. Distinct functional motifs within the IL-17 receptor regulate signal transduction and target gene expression. Proc Natl Acad Sci U S A. 2007;104:7506–7511. doi: 10.1073/pnas.0611589104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushima Y, Kikkawa Y, Takada T, Matsuoka K, Seki Y, Yoshida H, Minegishi Y, Karasuyama H, Yonekawa H. An atopic dermatitis-like skin disease with hyper-IgE-emia develops in mice carrying a spontaneous recessive point mutation in the Traf3ip2 (Act1/CIKS) gene. J Immunol. 2010;185:2340–2349. doi: 10.4049/jimmunol.0900694. [DOI] [PubMed] [Google Scholar]
- Mauro C, Vito P, Mellone S, Pacifico F, Chariot A, Formisano S, Leonardi A. Role of the adaptor protein CIKS in the activation of the IKK complex. Biochem Biophys Res Commun. 2003;309:84–90. doi: 10.1016/s0006-291x(03)01532-8. [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellett M, Atzei P, Horgan A, Hams E, Floss T, Wurst W, Fallon PG, Moynagh PN. Orphan receptor IL-17RD tunes IL-17A signalling and is required for neutrophilia. Nat Commun. 2012;3:1119. doi: 10.1038/ncomms2127. [DOI] [PubMed] [Google Scholar]
- Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, Kanno Y, Spalding C, Elloumi HZ, Paulson ML, et al. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature. 2008;452:773–776. doi: 10.1038/nature06764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minegishi Y, Saito M, Nagasawa M, Takada H, Hara T, Tsuchiya S, Agematsu K, Yamada M, Kawamura N, Ariga T, et al. Molecular explanation for the contradiction between systemic Th17 defect and localized bacterial infection in hyper-IgE syndrome. J Exp Med. 2009;206:1291–1301. doi: 10.1084/jem.20082767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monia O, Aydan I, Ilhan T, Figen D, Ithaisa S, Sigifredo P-S, Melike K, Gonul T, Chris N, Elena C, et al. Clinical features of candidiasis in patients with inherited interleukin-12 receptor β1 deficiency. 2013. submitted. [Google Scholar]
- Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nograles KE, Zaba LC, Shemer A, Fuentes-Duculan J, Cardinale I, Kikuchi T, Ramon M, Bergman R, Krueger JG, Guttman-Yassky E. IL-22-producing “T22” T cells account for upregulated IL-22 in atopic dermatitis despite reduced IL-17-producing TH17 T cells. J Allergy Clin Immunol. 2009;123:1244–1252. e1242. doi: 10.1016/j.jaci.2009.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novatchkova M, Leibbrandt A, Werzowa J, Neubuser A, Eisenhaber F. The STIR-domain superfamily in signal transduction, development and immunity. Trends Biochem Sci. 2003;28:226–229. doi: 10.1016/S0968-0004(03)00067-7. [DOI] [PubMed] [Google Scholar]
- Okada S, Cypowyj S, Liu L, Kong X-F, Kreins AY, Mekki N, Toubiana J, Hiller J, Okada C, Boisson B, et al. Gain-of-function STAT1 mutations underlying CMC: enhanced responses to IFN-α/β, IFN-γ, and IL-27 impair IL-17 T-cell immunity. 2013. Submitted. [Google Scholar]
- Onishi RM, Gaffen SL. Interleukin-17 and its target genes: mechanisms of interleukin-17 function in disease. Immunology. 2010;129:311–321. doi: 10.1111/j.1365-2567.2009.03240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owyang AM, Zaph C, Wilson EH, Guild KJ, McClanahan T, Miller HR, Cua DJ, Goldschmidt M, Hunter CA, Kastelein RA, Artis D. Interleukin 25 regulates type 2 cytokine-dependent immunity and limits chronic inflammation in the gastrointestinal tract. J Exp Med. 2006;203:843–849. doi: 10.1084/jem.20051496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prando C, Samarina A, Bustamante J, Boisson-Dupuis S, Cobat A, Picard C, Alsum Z, Al-Jumaah S, Al-Hajjar S, Frayha H, et al. Inherited IL-12p40 Deficiency: Genetic, Immunologic, and Clinical Features of 49 Patients From 30 Kindreds. Medicine (Baltimore) 2013;92:109–122. doi: 10.1097/MD.0b013e31828a01f9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puel A, Cypowyj S, Bustamante J, Wright JF, Liu L, Lim HK, Migaud M, Israel L, Chrabieh M, Audry M, et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science. 2011;332:65–68. doi: 10.1126/science.1200439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puel A, Cypowyj S, Marodi L, Abel L, Picard C, Casanova JL. Inborn errors of human IL-17 immunity underlie chronic mucocutaneous candidiasis. Curr Opin Allergy Clin Immunol. 2012;12:616–622. doi: 10.1097/ACI.0b013e328358cc0b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puel A, Doffinger R, Natividad A, Chrabieh M, Barcenas-Morales G, Picard C, Cobat A, Ouachee-Chardin M, Toulon A, Bustamante J, et al. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J Exp Med. 2010;207:291–297. doi: 10.1084/jem.20091983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Y, Giltiay N, Xiao J, Wang Y, Tian J, Han S, Scott M, Carter R, Jorgensen TN, Li X. Deficiency of Act1, a critical modulator of B cell function, leads to development of Sjogren’s syndrome. Eur J Immunol. 2008;38:2219–2228. doi: 10.1002/eji.200738113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Y, Liu C, Hartupee J, Altuntas CZ, Gulen MF, Jane-Wit D, Xiao J, Lu Y, Giltiay N, Liu J, et al. The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat Immunol. 2007;8:247–256. doi: 10.1038/ni1439. [DOI] [PubMed] [Google Scholar]
- Qian Y, Qin J, Cui G, Naramura M, Snow EC, Ware CF, Fairchild RL, Omori SA, Rickert RC, Scott M, et al. Act1, a negative regulator in CD40- and BAFF-mediated B cell survival. Immunity. 2004;21:575–587. doi: 10.1016/j.immuni.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Ramirez-Carrozzi V, Sambandam A, Luis E, Lin Z, Jeet S, Lesch J, Hackney J, Kim J, Zhou M, Lai J, et al. IL-17C regulates the innate immune function of epithelial cells in an autocrine manner. Nat Immunol. 2011;12:1159–1166. doi: 10.1038/ni.2156. [DOI] [PubMed] [Google Scholar]
- Renner ED, Rylaarsdam S, Anover-Sombke S, Rack AL, Reichenbach J, Carey JC, Zhu Q, Jansson AF, Barboza J, Schimke LF, et al. Novel signal transducer and activator of transcription 3 (STAT3) mutations, reduced T(H)17 cell numbers, and variably defective STAT3 phosphorylation in hyper-IgE syndrome. J Allergy Clin Immunol. 2008;122:181–187. doi: 10.1016/j.jaci.2008.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickel EA, Siegel LA, Yoon BR, Rottman JB, Kugler DG, Swart DA, Anders PM, Tocker JE, Comeau MR, Budelsky AL. Identification of functional roles for both IL-17RB and IL-17RA in mediating IL-25-induced activities. J Immunol. 2008;181:4299–4310. doi: 10.4049/jimmunol.181.6.4299. [DOI] [PubMed] [Google Scholar]
- Romberg N, Morbach H, Lawrence MG, Kim S, Kang I, Holland SM, Milner JD, Meffre E. Gain-of-function STAT1 mutations are associated with PD-L1 overexpression and a defect in B-cell survival. J Allergy Clin Immunol. 2013 doi: 10.1016/j.jaci.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampaio EP, Hsu AP, Pechacek J, Bax HI, Dias DL, Paulson ML, Chandrasekaran P, Rosen LB, Carvalho DS, Ding L, et al. Signal transducer and activator of transcription 1 (STAT1) gain-of-function mutations and disseminated coccidioidomycosis and histoplasmosis. J Allergy Clin Immunol. 2013;131:1624–1634. e1617. doi: 10.1016/j.jaci.2013.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Ullrich SJ, Zhang J, Connolly K, Grzegorzewski KJ, Barber MC, Wang W, Wathen K, Hodge V, Fisher CL, et al. A novel cytokine receptor-ligand pair. Identification, molecular characterization, and in vivo immunomodulatory activity. J Biol Chem. 2000;275:19167–19176. doi: 10.1074/jbc.M910228199. [DOI] [PubMed] [Google Scholar]
- Smeekens SP, Plantinga TS, van de Veerdonk FL, Heinhuis B, Hoischen A, Joosten LA, Arkwright PD, Gennery A, Kullberg BJ, Veltman JA, et al. STAT1 hyperphosphorylation and defective IL12R/IL23R signaling underlie defective immunity in autosomal dominant chronic mucocutaneous candidiasis. PLoS One. 2011;6:e29248. doi: 10.1371/journal.pone.0029248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Zhu S, Shi P, Liu Y, Shi Y, Levin SD, Qian Y. IL-17RE is the functional receptor for IL-17C and mediates mucosal immunity to infection with intestinal pathogens. Nat Immunol. 2011;12:1151–1158. doi: 10.1038/ni.2155. [DOI] [PubMed] [Google Scholar]
- Sun D, Novotny M, Bulek K, Liu C, Li X, Hamilton T. Treatment with IL-17 prolongs the half-life of chemokine CXCL1 mRNA via the adaptor TRAF5 and the splicing-regulatory factor SF2 (ASF) Nat Immunol. 2011;12:853–860. doi: 10.1038/ni.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takezaki S, Yamada M, Kato M, Park MJ, Maruyama K, Yamazaki Y, Chida N, Ohara O, Kobayashi I, Ariga T. Chronic mucocutaneous candidiasis caused by a gain-of-function mutation in the STAT1 DNA-binding domain. J Immunol. 2012;189:1521–1526. doi: 10.4049/jimmunol.1200926. [DOI] [PubMed] [Google Scholar]
- Uzel G, Sampaio EP, Lawrence MG, Hsu AP, Hackett M, Dorsey MJ, Noel RJ, Verbsky JW, Freeman AF, Janssen E, et al. Dominant gain-of-function STAT1 mutations in FOXP3 wild-type immune dysregulation-polyendocrinopathy-enteropathy-X-linked-like syndrome. J Allergy Clin Immunol. 2013;131:1611–1623. e1613. doi: 10.1016/j.jaci.2012.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Veerdonk FL, Plantinga TS, Hoischen A, Smeekens SP, Joosten LA, Gilissen C, Arts P, Rosentul DC, Carmichael AJ, Smits-van der Graaf CA, et al. STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N Engl J Med. 2011;365:54–61. doi: 10.1056/NEJMoa1100102. [DOI] [PubMed] [Google Scholar]
- Wang C, Wu L, Bulek K, Martin BN, Zepp JA, Kang Z, Liu C, Herjan T, Misra S, Carman JA, et al. The psoriasis-associated D10N variant of the adaptor Act1 with impaired regulation by the molecular chaperone hsp90. Nat Immunol. 2013a;14:72–81. doi: 10.1038/ni.2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Lin Z, Gao L, Wang A, Wan Z, Chen W, Yang Y, Li R. Exome sequencing reveals a signal transducer and activator of transcription 1 (STAT1) mutation in a child with recalcitrant cutaneous fusariosis. J Allergy Clin Immunol. 2013b;131:1242–1243. doi: 10.1016/j.jaci.2012.11.005. [DOI] [PubMed] [Google Scholar]
- Wells RS. Chronic oral candidiasis (autosomal recessive inheritance) (three cases) Proc R Soc Med. 1970;63:890–891. [PMC free article] [PubMed] [Google Scholar]
- Wells RS, Higgs JM, Macdonald A, Valdimarsson H, Holt PJ. Familial chronic muco-cutaneous candidiasis. J Med Genet. 1972;9:302–310. doi: 10.1136/jmg.9.3.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson DM. Chronic hyperplastic candidiasis and squamous carcinoma. Br J Dermatol. 1969;81:125–127. doi: 10.1111/j.1365-2133.1969.tb15992.x. [DOI] [PubMed] [Google Scholar]
- Zhang B, Liu C, Qian W, Han Y, Li X, Deng J. Crystal Structure of IL-17 Receptor B SEFIR Domain. J Immunol. 2013 doi: 10.4049/jimmunol.1202922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao A, Urban JF, Jr, Sun R, Stiltz J, Morimoto M, Notari L, Madden KB, Yang Z, Grinchuk V, Ramalingam TR, et al. Critical role of IL-25 in nematode infection-induced alterations in intestinal function. J Immunol. 2010;185:6921–6929. doi: 10.4049/jimmunol.1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.