

Abstract
Chronic mucocutaneous candidiasis disease (CMCD) is characterized by recurrent or persistent infections of the skin, nails, oral and genital mucosae caused by Candida albicans and, to a lesser extent, Staphylococcus aureus, in patients with no other infectious or autoimmune manifestations. We report two genetic etiologies of CMCD: autosomal recessive deficiency in the cytokine receptor, interleukin-17 receptor A (IL-17RA), and autosomal dominant deficiency of the cytokine IL-17F. IL-17RA deficiency is complete, abolishing cellular responses to IL-17A and IL-17F homo- and heterodimers. By contrast, IL-17-F deficiency is partial, with mutant IL-17F-containing homo- and heterodimers displaying impaired, but not abolished activity. These experiments of nature indicate that human IL-17A and IL-17F are essential for mucocutaneous immunity against C. albicans, but otherwise largely redundant.
Chronic mucocutaneous candidiasis (CMC) is characterized by infections of the skin, nails, oral and genital mucosae, with C. albicans, which is commensal in healthy individuals (1). In patients with inherited or acquired T-cell immunodeficiencies, CMC is associated with various infectious diseases (1). In patients with STAT3 deficiency and a lack of IL-17A- and IL-22-producing T cells (2-5), CMC is associated with severe cutaneous and pulmonary staphylococcal infections (1). In some patients with IL-12p40 or IL-12Rβ1 deficiency and mycobacterial disease (2), and in a family with CARD9 deficiency with systemic candidiasis and peripheral dermatophytosis (6), CMC and low proportions of IL-17A-producing T cells were also documented. Finally, CMC is the only infection of patients with AIRE deficiency, who have neutralizing autoantibodies against IL-17A, IL-17F, and IL-22 (7, 8). These data suggest that human IL-17A, IL-17F, and/or IL-22 are involved in mucocutaneous immunity to C. albicans (1). CMC disease (CMCD), the molecular and cellular basis of which is unknown, consists of CMC in the absence of other overt infectious or autoimmune signs (1). CMCD was initially thought to be benign, until squamous cell carcinoma (9) and cerebral aneurysms (10) were reported. First described in 1967 in sporadic cases (11), familial CMC segregating as autosomal dominant (AD) (12) and autosomal recessive (AR) traits (13) was soon reported. We thus searched for the genetic basis of CMCD, testing the hypothesis that CMCD may be caused by inborn errors of IL-17A, IL-17F, or IL-22 immunity (1, 14).
We first investigated a French child born to first-cousin parents of Moroccan descent (Fig. 1A, Report S1). He presented with C. albicans dermatitis during the neonatal period and displayed S. aureus dermatitis at five months of age. Known causes of CMC were excluded clinically and genetically and the lack of any phenotype other than CMC led to a diagnosis of AR CMCD. We sequenced the candidate genes encoding IL-22, IL-22RA1, IL-10RB, IL-17A, IL-17F, IL-17RA, and IL-17RC (15-17). IL-22 binds as a monomer to its receptor, composed of IL-22RA1 and IL-10RB, whereas IL-17A and IL-17F can form homo- or heterodimers that signal via a receptor comprising IL-17RA and IL-17RC chains. The child was found to be homozygous for the c.850C>T nonsense mutation (c.850C>T/c.850C>T), which replaces the glutamine codon in position 284 with a stop codon (Q284X/Q284X) in the IL17RA gene (Fig. 1B). This premature stop codon is located in the part of the gene encoding the extracellular domain of IL-17RA, upstream from the transmembrane domain sequence (Fig. 1C). No mutations were found elsewhere in IL17RA or in any of the other six genes sequenced. The parents and siblings of this child are healthy and heterozygous for the mutant allele, consistent with AR inheritance for this trait. The mutant allele was not found in 1,065 healthy controls from 52 ethnic groups from the CEPH-HGD panel, 100 French controls and 70 Moroccan controls of Berber descent, ruling out an irrelevant polymorphism and suggesting that the mutation may define a rare AR CMCD-causing allele.
Fig. 1.
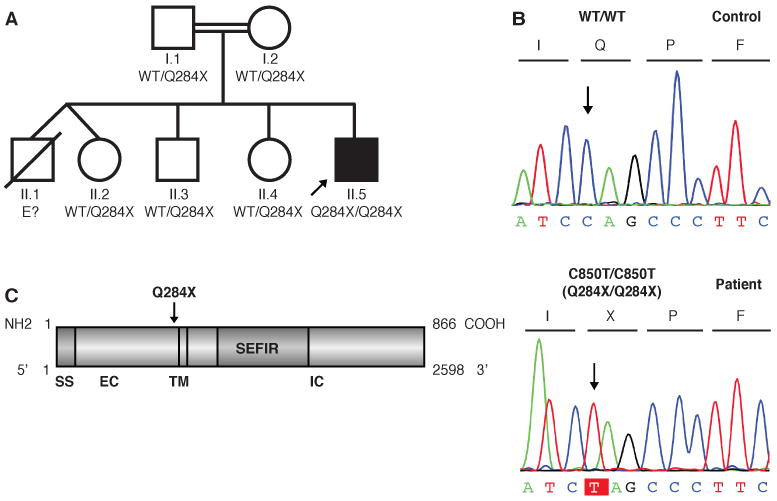
A kindred with autosomal recessive IL-17RA deficiency. (A) Pedigree of the family established by IL-17RA genotyping. The proband is indicated by an arrow. E? indicates individuals whose genetic status could not be evaluated. (B) IL17RA DNA sequence electrophoregrams for a control and the patient. (C) Schematic diagram of the IL-17RA protein with the signal sequence (SS), extracellular (EC), transmembrane (TM), intracellular (IC) and SEFIR (similar expression to FGF/IL-17R) domains, and the position within the extracellular domain affected by the mutation.
The IL-17RA protein was not detected on the surface of fibroblasts, peripheral blood mononuclear cells (PBMCs), or, more specifically, CD4+ T cells, CD8+ T cells and monocytes from the patient, as shown by flow cytometry with two specific antibodies against the extracellular domain (Fig. 2A, Fig. S1). The absence of IL-17RA had no impact on the expression of IL-17RC, which was normal on the patient's monocytes (the only leukocyte subset expressing IL-17RC in controls) and fibroblasts (Fig. S1 and S2). Likewise, IL-22RA1 was normally expressed on the patient's fibroblasts (Fig. S2). The patient also had a normal proportion of circulating IL-17A- and IL-22- producing T cells (Fig. S3). We investigated whether the lack of IL-17RA expression had any functional consequences for the response to IL-17 cytokines, by testing the responses of the patient's fibroblasts to various concentrations of recombinant IL-17A and IL-17F homodimers, and to IL-17A/IL-17F heterodimers (16, 17). Like NEMO-deficient fibroblasts, which have impaired NF-κB activity and unlike fibroblasts from a healthy control, the patient's fibroblasts did not respond to any of the three IL-17 cytokines, in terms of IL-6 and growth-regulated oncogene-α (GRO-α) induction (18), as assessed by ELISA on supernatants (Fig. 2B, and C). Moreover, the patient's PBMCs did not respond above baseline to IL-17A or IL-17F for any of the cytokines tested (Fig. S4A). Transfection of the patient's fibroblasts with wild-type (WT) IL17RA, but not with a mock vector, restored IL-17RA expression and the response to IL-17 cytokines (Fig. 2, D-F). By contrast, IL-6 production by NEMO-deficient cells was not rescued by transfection with IL17RA (Fig. S4B). Thus, the patient with CMCD studied displayed AR, complete IL-17RA deficiency and a lack of cellular responses to at least three IL-17 cytokine dimers — IL-17A, IL-17F, and IL-17A/IL-17F — in fibroblasts and leukocytes.
Fig. 2.
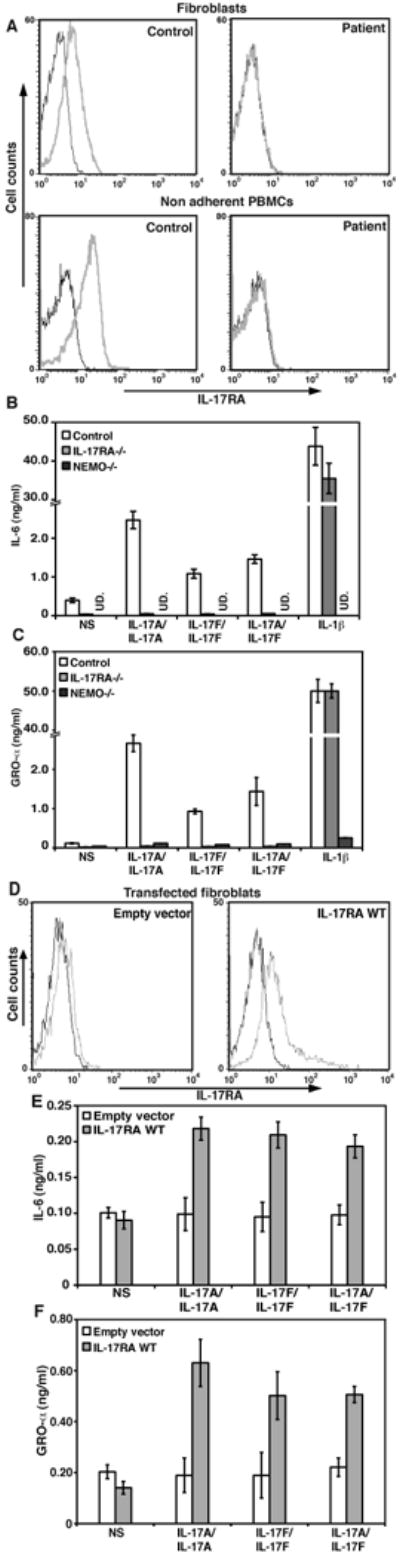
Production and function of the mutant IL-17RA chain. (A) IL-17RA expression in SV40-immortalized fibroblasts from a control and the patient (top panel) and non adherent PBMCs (lower panel), as detected by flow cytometry. Isotype control (black); IL-17RA antibody (gray). (B) IL-6 and (C) GRO-α production by SV40-immortalized fibroblasts from a control, the patient, and NEMO-deficient cells after 24 hours of stimulation with IL-17A, IL-17F and IL-17A/IL-17F, mean +/- standard deviation (SD) error bars of three independent experiments, as detected by ELISA (UD. Undetectable). (D) IL-17RA expression in SV40-immortalized fibroblasts from the patient, transfected with the empty pORF9mcs plasmid (left panel) or the pORF9-hIL-17RA plasmid (right panel), as detected by flow cytometry. Isotype control (black); IL17RA antibody (gray). (E) IL-6 and (F) GRO-α production by SV40-immortalized fibroblasts from the patient, transfected with the empty pORF9mcs plasmid (white) or the pORF9-hIL17RA plasmid (gray), after 24 hours of stimulation with IL-17A, IL-17F and IL-17A/IL-17F, mean +/- SD error bars of three independent experiments, as detected by ELISA.
We then investigated a multiplex family from Argentina, with AD inheritance of CMCD (Fig. 3A, Report S2). The IL22, IL22RA, IL10RB, IL17RA, IL17RC and IL17A genes contained no mutations, but a heterozygous missense mutation was found in the IL17F gene of the index case. This mutation, c.284C>T, replaced the serine residue in position 65 of the mature protein with a leucine residue (S65L) (Fig. 3, B and C). The S65 residue is conserved across mammalian species (Fig. S5). Moreover, the sequencing of 1,074 control individuals from the CEPH-HGD panel ruled out the possibility that this mutation was an irrelevant polymorphism. Computational analysis showed that S65 lies in the cavity of the protein, which is thought to be involved in cytokine-receptor binding (Fig. 3C) (19). No other IL17F variations were found in the index case, including the IL17F g.7488T>C (rs763780) polymorphism, replacing a histidine with an arginine residue in position 161 of the protein (H161R), a mutation previously thought to be loss-of-function (20). By contrast, we found that the H161R allele encoded an IL-17F protein able to stimulate murine lung epithelial cells (MLE) (Fig. S6). Heterozygosity for the S65L allele was found in all members of the kindred with CMCD tested; we were unable to genotype the fifth patient (III.1 in Fig. 3A), who died at six years of age from complications of the disease. The mutant allele was found in only two apparently healthy family members, aged nine months (III.3 in Fig. 3A) and 21 years (II.8 in Fig. 3A), suggesting incomplete clinical penetrance. We did not detect IL-17F-expressing T cells in controls by flow cytometry, but the patients tested displayed normal proportions of IL-17A- and IL-22-expressing T cells and their PBMCs secreted normal amounts of cytokines, as measured by Bioplex (Fig. S7, A and B).
Fig. 3.
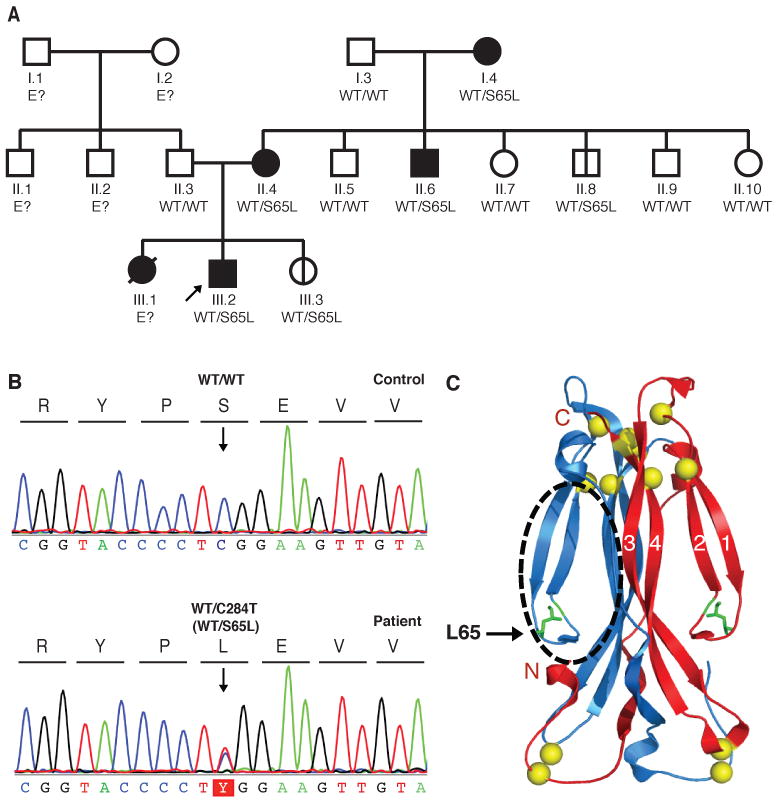
A kindred with autosomal dominant IL-17F deficiency. (A) Family pedigree, with allele segregation. The patients, shown in black, are all heterozygous for the mutation, as is II.8, who is asymptomatic. The proband is indicated by an arrow. E? indicates individuals whose genetic status could not be evaluated. III.3 is a nine-month-old baby, also heterozygous for the mutation and currently asymptomatic. All other family members are healthy and wild-type for IL17F and are shown in white. (B) Heterozygous c.284C>T mutation in the patients. IL17F DNA sequence electrophoregrams of a control and the patient III.2. (C) Ribbon trace of the IL-17F dimer. Beta strands are labeled. Sulfur atoms are shown in yellow. The position of the cavity that binds to the receptor is indicated by a black circle.
We investigated the possible deleterious effects of the S65L mutation, by producing the mutant IL-17F protein in HEK293 cells. The mutation did not seem to affect production of the monomeric protein or the formation of IL-17F homodimers (mutant/mutant and wild-type/mutant) or heterodimers with IL-17A (Fig. S8). The mutant-containing dimers seemed to bind normally to monomeric IL-17 receptors (IL-17RA and IL-17RC), as shown by surface plasmon resonance (Table S1, Fig. S9). However, the mutant proteins did not bind IL-17RA on fibroblasts, as shown by flow cytometry, with IL-17RA-deficient cells as controls (confirming that their lack of IL-17RA expression prevented cytokine binding) (Fig. S10, S11). Accordingly, when control fibroblasts (Fig 4, A and B) and keratinocytes (Fig. S12, A and B) were stimulated with mutant S65L IL-17F homodimers, they displayed much weaker IL-6 and GRO-α induction than observed with WT IL-17F homodimers (IL-17WT), IL-17A homodimers, or IL-17A/IL-17FWT heterodimers (18). Moreover, control PBMCs showed impaired induction of several cytokines when stimulated with S65L IL-17F homodimers compared to WT IL-17F homodimers (Fig. S12C). These data suggest that the IL17F S65L allele is severely hypomorphic (Fig. 4, A and B, Fig. S12, A and B). Furthermore, when the S65L mutant IL-17F formed a heterodimer with either IL-17FWT or IL-17A, the induction of IL-6 and GRO-α was severely impaired in control fibroblasts (Fig. 4, A and B) and keratinocytes (Fig. S12, A-B), indicating a dominant-negative effect of this allele. Finally, as predicted by the lack of binding of mutant cytokine dimers to their receptor (Fig. S11), these dimers did not compete with WT dimers (Fig. S13, A-D). Thus, the AD CMCD in this kindred results from a hypomorphic, dominant-negative IL17F allele, which impairs the receptor-binding and bioactivity of both IL-17F homodimers and IL-17A/IL-17F heterodimers.
Fig. 4.

Function of the mutant IL-17F protein. (A) Production of IL-6 and (B) GROα by control SV-40 fibroblasts in response to increasing doses (ng/ml) of IL-17A, IL-17F wild-type (IL-17FWT), mutant IL-17F (IL-17FS65L), IL-17FWT/IL-17FS65L homodimers and of IL-17A/IL-17FWT and IL-17A/IL-17FS65L heterodimers for 24 hours, mean +/- SD error bars of three independent experiments, as detected by ELISA.
IL-17RA and IL-17F deficiency underlying mucocutaneous disease caused by C. albicans and, to a lesser extent, S. aureus, is consistent with the mouse model (21). IL-17RA- and IL-17RC-deficient mice were more susceptible to oropharyngeal candidiasis (22, 23) and IL-17RA-deficient mice to cutaneous staphylococcal disease (24). IL-17A-deficient mice also display impaired clearance of C. albicans skin infection (25). IL-17F-deficient mice have not yet been tested, but IL-23-deficient mice with impaired expression of IL-17A and IL-17F are also vulnerable (25). IL-17A or IL-17F alone are not required for peripheral immunity to S. aureus, but mice deficient for both IL-17A and IL-17F display an impaired peripheral immunity to S. aureus (26). Somewhat at odds with our observations, IL-17A is also required for systemic immunity to C. albicans (27) and S. aureus (28). Moreover, mice with IL-17RA, IL-17RC, IL-17A, or IL-17F deficiency are vulnerable to multiple infections at various anatomical sites (21, 28). Overall, our report indicates that human IL-17A and IL-17F are essential for protective immunity to C. albicans and, to a lesser extent, S. aureus in the nails, skin, oral and genital mucosae, but otherwise redundant. We cannot exclude the possibility that other infections may occur in patients with inborn errors of IL-17 immunity. In any event, in natura, inborn errors of IL-17 immunity clearly impair mucocutaneous immunity to Candida albicans (29). Patients receiving IL-17 blocking agents should be carefully monitored, at least for mucocutaneous infections (30).
Supplementary Material
Acknowledgments
We thank the patients, their families, and their clinicians. We also thank all members of the laboratory for helpful discussions, especially T. Kochetkov for keratinocyte culture. This work was supported by institutional funding from INSERM, University Paris Descartes, the Rockefeller University, The Rockefeller University CTSA grant number 5UL1RR024143-04, the St. Giles Foundation, and the Candidoser association awarded to JLC. Genomic DNA sequences of the mutations IL-17RA and IL-17F can be found in GenBank.
Footnotes
This manuscript has been accepted for publication in Science. This version has not undergone final editing. Please refer to the complete version of record at http://www.sciencemag.org/. The manuscript may not be reproduced or used in any manner that does not fall within the fair use provisions of the Copyright Act without the prior, written permission of AAAS.
References and Notes
- 1.Puel A, et al. Curr Opin Immunol. 2010 Aug;22:467. doi: 10.1016/j.coi.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Beaucoudrey L, et al. J Exp Med. 2008 Jul 7;205:1543. doi: 10.1084/jem.20080321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ma CS, et al. J Exp Med. 2008 Jul 7;205:1551. doi: 10.1084/jem.20080218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Minegishi Y, et al. J Exp Med. 2009 Jun 8;206:1291. doi: 10.1084/jem.20082767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Milner JD, et al. Nature. 2008 Apr 10;452:773. [Google Scholar]
- 6.Glocker EO, et al. N Engl J Med. 2009 Oct 29;361:1727. doi: 10.1056/NEJMoa0810719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kisand K, et al. J Exp Med. 2010 Feb 15;207:299. doi: 10.1084/jem.20091669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Puel A, et al. J Exp Med. 2010 Feb 15;207:291. doi: 10.1084/jem.20091983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Williamson DM. Br J Dermatol. 1969 Feb;81:125. doi: 10.1111/j.1365-2133.1969.tb15992.x. [DOI] [PubMed] [Google Scholar]
- 10.Leroy D, Dompmartin A, Houtteville JP, Theron J. Dermatologica. 1989;178:43. doi: 10.1159/000248386. [DOI] [PubMed] [Google Scholar]
- 11.Chilgren RA, Quie PG, Meuwissen HJ, Hong R. Lancet. 1967 Sep 30;2:688. doi: 10.1016/s0140-6736(67)90974-9. [DOI] [PubMed] [Google Scholar]
- 12.Canales L, Middlemas RO, 3rd, Louro JM, South MA. Lancet. 1969 Sep 13;2:567. doi: 10.1016/s0140-6736(69)90264-5. [DOI] [PubMed] [Google Scholar]
- 13.Wells RS, Higgs JM, Macdonald A, Valdimarsson H, Holt PJ. J Med Genet. 1972 Sep;9:302. doi: 10.1136/jmg.9.3.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Casanova JL, Abel L. Science. 2007 Aug 3;317:617. doi: 10.1126/science.1142963. [DOI] [PubMed] [Google Scholar]
- 15.Wolk K, Witte E, Witte K, Warszawska K, Sabat R. Semin Immunopathol. 2010 Mar;32:17. doi: 10.1007/s00281-009-0188-x. [DOI] [PubMed] [Google Scholar]
- 16.Korn T, Bettelli E, Oukka M, Kuchroo VK. Annu Rev Immunol. 2009;27:485. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 17.Gaffen SL. Nat Rev Immunol. 2009 Aug;9:556. doi: 10.1038/nri2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wright JF, et al. J Immunol. 2008 Aug 15;181:2799. doi: 10.4049/jimmunol.181.4.2799. [DOI] [PubMed] [Google Scholar]
- 19.Hymowitz SG, et al. Embo J. 2001 Oct 1;20:5332. doi: 10.1093/emboj/20.19.5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kawaguchi M, et al. J Allergy Clin Immunol. 2006 Apr;117:795. doi: 10.1016/j.jaci.2005.12.1346. [DOI] [PubMed] [Google Scholar]
- 21.Khader SA, Gaffen SL, Kolls JK. Mucosal Immunol. 2009 Sep;2:403. doi: 10.1038/mi.2009.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Conti HR, et al. J Exp Med. 2009 Feb 16;206:299. doi: 10.1084/jem.20081463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ho AW, et al. J Immunol. 2010 Jul 15;185:1063. doi: 10.4049/jimmunol.0903739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cho JS, et al. J Clin Invest. 2010;120:1762. doi: 10.1172/JCI40891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kagami S, Rizzo HL, Kurtz SE, Miller LS, Blauvelt A. J Immunol. 2010 Nov 1;185:5453. doi: 10.4049/jimmunol.1001153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ishigame H, et al. Immunity. 2009 Jan 16;30:108. doi: 10.1016/j.immuni.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 27.Saijo S, et al. Immunity. 2010 May 28;32:681. doi: 10.1016/j.immuni.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 28.Henningsson L, et al. Infect Immun. 2010 Jun 28; doi: 10.1128/IAI.00385-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alcais A, et al. Ann N Y Acad Sci. 2010 Dec;1214:18. doi: 10.1111/j.1749-6632.2010.05834.x. [DOI] [PubMed] [Google Scholar]
- 30.Hueber W, et al. Sci Transl Med. 2010 Oct 6;2:52ra72. doi: 10.1126/scitranslmed.3001107. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.