

Abstract
An emerging treatment option for chronic lymphocytic leukemia (CLL) is to make cytotoxic immune cells express a chimeric antigen receptor (CAR) that recognizes specific surface molecules on CLL cells. Here an mRNA coding for an anti-CD19 CAR was transfected into the NK-92 cell line by electroporation. In contrast to cDNA, mRNA resulted in high transfection efficiency (47.2 ± 8% versus <5% for cDNA) with minimal effect on cell viability. NK-92 cells expressing anti-CD19 CAR killed previously resistant CD19+ BALL cell lines, as well as primary CLL cells and therefore may present a safe, cell-based, targeted treatment for patients with CLL.
Keywords: CLL, NK cells, Cytotoxicity, Transfection
1. Introduction
Chronic lymphocytic leukemia (CLL) is the most common leukemia in the western world. Although it responds to chemotherapy, the disease usually recurs with resistance developing over time. Combining chemotherapy with monoclonal antibodies directed against specific surface markers, such as CD20, CD22 and CD23, seems to improve the outcome by providing an alternative way of inducing apoptosis in clonal cells [1,2]. Another treatment modality, which is not cross-reactive with chemotherapy and monoclonal antibody treatment, is to make use of the cytotoxicity mediated by immune cells such as T-cells and NK cells that release perforin and granzymes, inducing target cell apoptosis by DNA degradation. The problem with cell therapy for hematological malignancies, though, is the lack of target specificity of these cells. Furthermore, autologous NK cells are generally blocked through activation of killer inhibitory receptors (KIR) by self-MHC antigens [3,4]. This inhibitory mechanism can be overcome either by using allogeneic or autologous NK cells whose KIR receptors have been blocked [5–7]. An alternative way to override this inhibition and make NK cells recognize malignant targets is to genetically engineer them to express activating receptors that recognize specific antigens on the tumor surface, thereby triggering the release of cytotoxic molecules. Chimeric antigen receptors (CAR) have recently received attention as one mode of retargeting cytotoxic cells [8–11]. These CAR consist of a single chain Fv fragment from an antibody specific for a surface antigen, linked to components of the T-cell receptor complex (such as CD3ζ) that can activate a cytolytic response through intracellular signaling molecules such as Syk/Zap70.
CLL cells constitutively express the CD19 antigen, which is a suitable target for CAR. Previous results have shown that the natural killer cell line NK-92 [12], although cytolytic to a wide range of tumor cells, may be unable to kill clonal cells of lymphoid origin [13]. However, it can be rendered cytotoxic against acute lymphoblastic leukemia (ALL) targets (both cell lines and primary patient cells) after they have been transfected with a retroviral vector coding for a murine CAR directed against human CD19 (αCD19-CAR) [14] or CD20 [11]. Despite their high transduction efficiency, retroviral constructs carry the risk of insertional mutagenesis and are for that reason not preferred for treatment in humans. The non-modified NK-92 cell line has already completed phase I trials [15,16] and this work was intended to introduce a non-viral αCD19-CAR expression vector that would be clinically useful for extending the indications for NK-92 treatment. Two different vector systems, based on DNA or mRNA constructs for GFP and αCD19-CAR, were tested for transfection efficiency and function in NK-92 using electroporation. Our results show that NK-92 cells can be effectively transfected with mRNA for αCD19-CAR, rendering them highly cytolytic towards previously resistant CD19 positive cell lines and primary CLL cells in vitro.
2. Material and methods
2.1. Cell lines, primary cells and culture conditions
K562 (CML in blast crisis, CD19−, NK-92 sensitive), REH, and SUP-B15 (both acute B-precursor ALL, CD19+, NK-92 resistant) cell lines were purchased from American Type Culture Collection (ATCC, Rockville, MD). SR-91 cell line (B-ALL, CD19−, NK-92 resistant) has been described before [17]. All cells were maintained in RPMI-1640 (Cambrex, Walkersville, MD) supplemented with 20% FBS (Cambrex) and with antibiotics: Penicillin 100 μl ml−1, Streptomycin 10 μg ml−1, and Amphotericin B 250 μg ml−1 (Gibco Invitrogen, Carlsbad, CA), and Ciprofloxacin 10 μg ml−1 (Mediatech, Herndon, VA). NK-92 cell line was maintained in Myelocult (StemCell Technology, Vancouver, BC) supplemented with recombinant human IL-2 (500 IU ml−1; Chiron, Emeryville, CA). Primary CLL cells were obtained from blood samples of untreated patients, after informed consent. CLL mononuclear cells were isolated by density gradient centrifugation using Ficoll–Hypaque Plus (Amersham Biosciences, Piscataway, NJ). CLL mononuclear cells were kept overnight in RPMI-1640 20% FBS at 4 °C, or frozen at −80 °C in 10% DMSO and thawed just before the cytotoxicity assays. Under both conditions spontaneous lysis was low (see Section 3).
2.2. Cloning of αCD19-CAR constructs
The αCD19-CAR construct in vector pLXSN [14] has been designed for production of retrovirus, but was not suitable for in vitro mRNA synthesis. The αCD19-CAR sequence was therefore amplified by PCR using a high accuracy Phusion DNA polymerase (New England Biolabs, Ipswich, MA) and the following primers: sense 5′-TTC GAC CCC GCC TCG ATC CTC C-3′, antisense 5′-GGA TCT GCC CTC GAG GGA GAA ATG CCC-3′; in order to introduce the XhoI restriction site (underlined) downstream of the sequence stop codon. The PCR product was cloned into vector pXT7 (a gift from Dr. Sergei Sokol, Mount Sinai School of Medicine, New York, together with plasmid pXT7-GFP), at the restriction sites EcoRI (present in the original CAR sequence) and XhoI. The plasmid pXT7 has a T7 promoter used to initiate mRNA synthesis, as well as the 5′- and 3′-untranslated regions (UTR) of human globin gene. These UTR provide a 3′ poly-adenylation sequence and enhance the stability of the mRNA in vivo (Fig. 1). For electroporation of αCD19-CAR DNA, the αCD19-CAR sequence was cloned into mammalian expression vector pcDNA™ 3.1 (Invitrogen, Carlsbad, CA) using the Directional TOPO Expression Kit (Invitrogen) according to the manufacturer’s instructions. Every new constructs was verified by DNA sequencing.
Fig. 1.
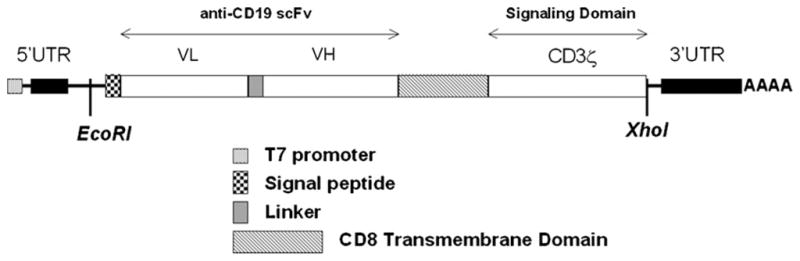
Schematic representation of the CD19-CAR mRNA. VH and VL: extracellular single strand antibody domains. 5′ and 3′ UTR are from human globin and enhance mRNA stability as well as provide a poly-adenylation sequence.
2.3. mRNA synthesis and electroporation
Cesium chloride preparation of plasmid pCMV-GFP (a gift from Dr. Lidija Covic, Tufts Medical Center, Boston) was used for electroporation of GFP DNA. For electroporation of mRNA, the plasmids pXT7-GFP and pXT7-αCD19-CAR were treated by the restriction enzyme Sal-I, which cuts downstream of the 3′ poly-adenylation sequence, and the linearized products were used as templates for in vitro mRNA synthesis reaction (T7 Ultra! mMessage mMachine kit, Ambion Applied Biosystems, Austin, TX) according to the manufacturer’s instructions. This kit couples T7-initiated transcription with poly-adenylated tail elongation in order to further increase mRNA stability and translation. Yield was determined by spectrophotometric dosage, and the integrity of the final mRNA products was checked by gel electrophoresis.
NK-92 cells were washed and resuspended in serum-free MEM medium (Gibco Invitrogen) at a concentration of 8 × 106 cells ml−1 (we observed that presence of serum caused degradation of mRNA). Cells were transferred into 4 mm electroporation cuvettes (Biorad, Hercules, CA) under the following conditions: 2 × 106 cells in 250 μl MEM mixed with DNA (20 μg ml−1 for GFP or αCD19-CAR), mRNA (40 μg ml−1 for GFP or 120 μg ml−1 for αCD19-CAR), or nothing. Electroporation was performed using a GenePulser II (Biorad), under the following conditions: 300 V, 150 μF, 200 Ω. Cells were immediately transferred into Myelocult medium and cultured at 37 °C in a 5% CO2 incubator. Levels of expression of GFP and αCD19-CAR proteins were monitored by flow cytometry using a Cyan flow cytometer (Dako, Carpinteria, CA). αCD19-CAR was detected using a biotinylated anti-mouse F(ab′)2 antibody (Jackson ImmunoResearch, West Grove, PA) and Allophycocyanin (APC)-conjugated Streptavidin (BD Biosciences, San Jose, CA).
2.4. Irradiation of NK-92 cells
Since any clinical application would require that NK-92 are irradiated prior to infusion into the patient, NK-92 cells were treated with a gamma radiation dose of 10 Gy (Shepherd Mark I gamma irradiator employing a 137Cs source, Tufts Medical Center), either 4 h before or 20 h after electroporation.
2.5. Cytotoxicity assays
Assays were performed as previously published [11,18]. Briefly, target cells (K562, SR-91, REH, or SUP-B15) were stained with the fluorescent dye PKH67-GL (Sigma–Aldrich, Saint Louis, MO) according to manufacturer’s instructions. Targets and effectors (i.e. electroporated NK-92 cells) were then combined at different effector to target ratios (E:T of 1:1, 2:1, 5:1 and 10:1) in a 96-well plate (Falcon BD, Franklin Lakes, NJ), briefly centrifuged, and incubated in RPMI-1640 20% FBS culture medium at 37 °C for 4 h in a 5% CO2 incubator. After incubation, cells were stained with propidium iodide (PI, Sigma–Aldrich) at 10 μg ml−1 in Ca2+/Mg2+-free phosphate buffer saline and analyzed immediately by flow cytometry. Dead target cells were identified as double positive for PKH67-GL and PI. Target cells and effector cells were also stained separately with PI to assess spontaneous cell lysis. The percentage of NK-mediated cytotoxicity was obtained by subtracting the percentage of PKH(+)/PI(+) cells for target cells alone (spontaneous lysis) from the percentage of PKH(+)/PI(+) cells in the samples with effectors.
3. Results
3.1. Comparison of DNA with RNA electroporation
For the production of αCD19-CAR mRNA a suitable vector was designed as schematically shown in Fig. 1. NK-92 cells were electroporated either with plasmid DNA (pCMV-GFP or pcDNA-αCD19-CAR, 25 μg ml−1), with mRNA (GFP or αCD19-CAR, respectively, 40 μg ml−1 and 120 μg ml−1), or without any nucleic acid (negative control). As shown in Fig. 2, electroporation with mRNA did not cause additional cell death compared to control (10.6 ± 3.3% (n = 7), 16.9 ± 3.4% (n = 7), 18.2 ± 5.1% (n = 10), respectively, for GFP, αCD19-CAR, and control) even with mRNA amounts of 120 μg ml−1. In contrast, electroporation with plasmid DNA caused two to three times more cell death than control: 38.7 ± 9.6% for GFP (n = 6) and 60.7 ± 5.8% for αCD19-CAR (n = 3).
Fig. 2.
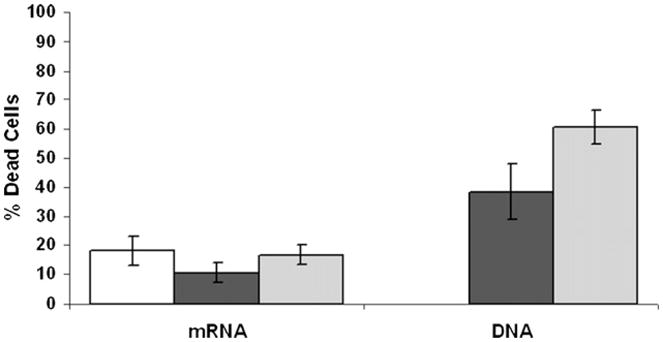
Viability of cells after electroporation. NK-92 cells were stained with PI 24 h after electroporation with GFP (■) or αCD19-CAR ( ), either in the form of DNA or mRNA, or without any nucleic acid (□, control).
), either in the form of DNA or mRNA, or without any nucleic acid (□, control).
3.2. Expression of GFP and αCD19-CAR proteins
Levels of expression of GFP and αCD19-CAR proteins in electroporated NK-92 cells were determined over time. As shown in Fig. 3A, at 24 h after electroporation 76 ± 4.9% (n = 8) of cells transfected with GFP mRNA expressed GFP protein, compared to only 2.1 ± 0.7% (n = 7) of the cells transfected with GFP DNA. Similarly, 47.2 ± 8% (n = 7) of cells transfected with αCD19-CAR mRNA expressed the CAR protein on their surface, whereas no detectable expression was seen in cells transfected with αCD19-CAR DNA (n = 3). Expression of GFP protein was stable over time, up to 3 days, and detectable for up to 5 days. αCD19-CAR was significantly less stable, with a peak of expression at around 24 h but no detectable levels after 3 days. In both cases, expression of the proteins was rapid, and occurred within the first 3 h after electroporation (Fig. 3B).
Fig. 3.

Expression and stability of the exogenous proteins. (A) Right: FACS plots of a representative experiment showing GFP expression (FITC), αCD19-CAR expression (APC), and PI staining (PE) following mRNA electroporation. Left: Expression levels of GFP (■) or αCD19-CAR () proteins at 24 h post-electroporation in NK-92 cells electroporated with DNA or mRNA, shown in percentage of the whole cell population. (B) Expression of GFP (●) or αCD19-CAR ( ) proteins over time in hours in NK-92 cells electroporated with mRNA.
) proteins over time in hours in NK-92 cells electroporated with mRNA.
3.3. NK-92 expressing αCD19-CAR are cytotoxic against previously resistant CD19-expressing cell lines
In order to determine whether electroporation and surface expression of CAR changed the cytolytic properties of NK-92 cells and whether the expressed αCD19-CAR was functional, we tested non-electroporated cells, control-electroporated cells, and GFP or αCD19-CAR-expressing cells (effectors) against the CD19− cell lines K562 and SR-91 as well as the CD19+ cell lines REH and SUP-B15 (targets). NK-92 is known to kill K562 target cells consistently even at low effector to target ratios [19]. As shown in Fig. 4, all types of effector cells killed K562 to a similar extent (53.5 ± 9.2% for non-electroporated (n = 5), 65.8 ± 4.6% for control (n = 3), 66.1 ± 0.1% for GFP (n = 2), and 61.7 ± 4.4% for αCD19CAR (n = 4) at E:T of 5:1), showing that electroporation did not significantly alter the cytolytic properties of the NK-92 cell line. B-ALL cell lines were resistant (<15%) to control-electroporated NK-92 cells and GFP-electroporated NK-92, as well as non-electroporated NK-92. In contrast, REH and SUP-B15 cell lines became highly sensitive to NK-92 expressing αCD19-CAR (76.3 ± 4.7% (n = 5) and 75.9 ± 7.3% (n = 4) respectively, p < 0.0001, at E:T of 5:1), even for lower E:T ratios (70.4% for REH, 68.5% for SUP-B15, p < 0.01, at E:T of 2:1). However, SR-91 cells were still resistant to NK-92 cells expressing αCD19-CAR, suggesting that the effect of αCD19-CAR is dependent on expression of CD19 by the target. NK-92 cells electroporated with both GFP and αCD19-CAR mRNA had the same properties as cells that received only one type of mRNA, suggesting that cells can express multiple functional constructs simultaneously (data not shown).
Fig. 4.
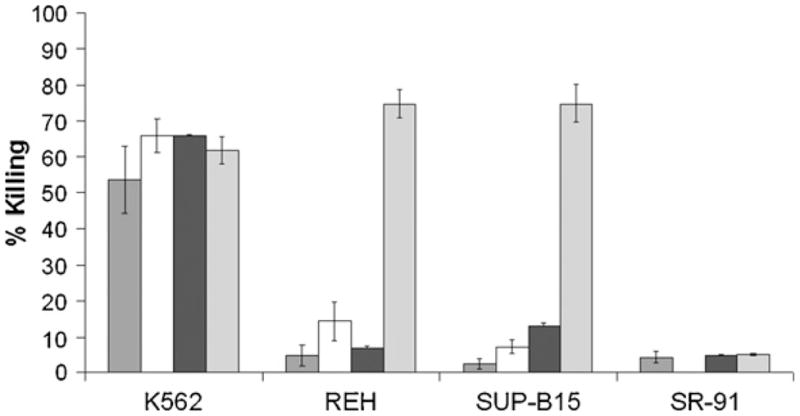
Cytolytic properties of electroporated NK-92 cells against CD19+ cell lines. Cytolytic efficiency, expressed in percentage of killed target cells, of NK-92 non-electroporated ( ), and electroporated without nucleic acid (□), with GFP mRNA (■), or with αCD19-CAR mRNA (), against cell lines K562, REH, SUP-B15, and SR-91. The data correspond to an effector to target ratio of 5:1.
), and electroporated without nucleic acid (□), with GFP mRNA (■), or with αCD19-CAR mRNA (), against cell lines K562, REH, SUP-B15, and SR-91. The data correspond to an effector to target ratio of 5:1.
3.4. Expression of αCD19-CAR in NK-92 restores killing of primary B-CLL cells
As shown in Fig. 5, control-electroporated NK-92 cells were only minimally cytolytic against primary CLL cells (<10% cell lysis). In contrast, NK-92 cells expressing αCD19-CAR killed primary CLL cells very effectively (>70% cell lysis). As observed for REH and SUP-B15 cell lines, lysis of CLL mediated by NK-92 cells expressing αCD19-CAR reached maximum levels for E:T ratios as low as 1 to 1 (data not shown). In all cases spontaneous lysis of CLL cells was low (4.6%, 3%, 2.4%, 4.3%, 4.5% for samples 1–5, respectively).
Fig. 5.
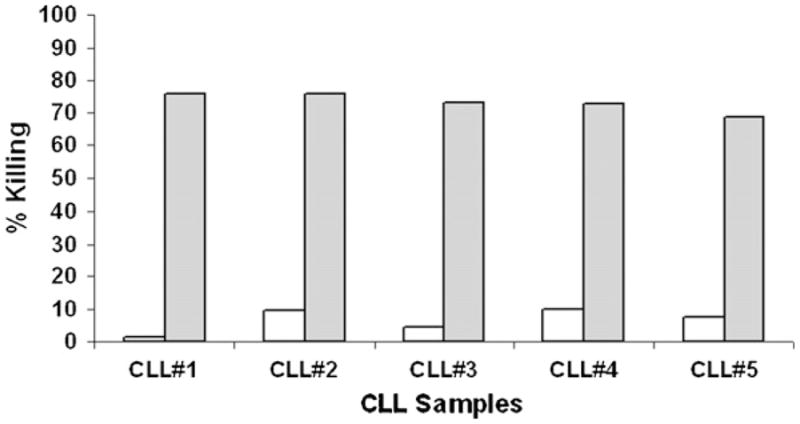
Cytolytic properties of electroporated NK-92 cells against CLL cells. Cytotoxicity, expressed in percentage of killed target cells, of NK-92 electroporated without nucleic acid (□), or with αCD19-CAR mRNA (), against five primary CLL MNC samples. The data are for an effector to target ratio of 5:1.
3.5. Gamma irradiated NK-92 αCD19-CAR cells maintain their specificity and cytotoxic properties
For any clinical application, NK-92 should be γ-irradiated prior to infusion into patients in order to prevent in vivo cell proliferation. In order to determine whether sub-lethal irradiation affects CAR-mediated cytotoxicity, we irradiated NK-92 cells either 4 h before or 20 h after electroporation, and tested their cytolytic activity at 20 h post-electroporation against K562 (n = 3), REH (n = 2), and SUP-B15 (n = 2) cell lines. As shown in Fig. 6, all NK-92 cells expressing αCD19-CAR were able to kill REH and SUP-B15 significantly better than control-electroporated ones (~70% compared to ~10%, p < 0.05), showing that irradiation did not impair expression and functionality of αCD19-CAR. We also observed that the cytotoxicity of NK-92 that were irradiated before electroporation (4 h) was consistently decreased compared to cells irradiated after electroporation (20 h), whether they express αCD19-CAR or not.
Fig. 6.

Effects of irradiation on cytolytic properties of electroporated NK-92 cells. Cytotoxicity, expressed in percentage of killed target cells, of NK-92 against cell lines K562, REH, and SUP-B15 for an effector to target ratio of 5:1. NK-92 cells were electroporated without nucleic acid (control, full box color) or with αCD19-CAR mRNA (stripes), and irradiated either 4 h prior to electroporation (■) or 20 h after electroporation ().
4. Discussion
Treatment for CLL is indicated in symptomatic patients with progression of the disease. Although purine analogs have been shown to control disease to a certain extent, they have not been shown to prolong survival [20]. In addition, resistance develops and remissions become shorter. More recently, Campath™, Rituximab™ and other monoclonal antibodies have been added to the “backbone” therapy with purine analogs with some improvement of outcome [21,22]. However, these “bio-chemotherapies” significantly compromise the immune system of the patient by inhibiting T- and B-lymphocyte function.
Targeted cytotoxic cell therapy has already been shown to result in control of viral infections [23]. More recently, allogeneic NK cells have been able to induce remissions in some patients with acute leukemia [24]. However, some tumors may display resistance mechanisms even against allogeneic NK cells. The development of CAR-expressing NK cells is one way to overcome this resistance, as it has been shown for the NK-92 cell line expressing a retroviral CAR construct recognizing ErbB2/HER2 on breast cancer cells [10] or CD20 on lymphoid cells [11]. The unmodified NK-92 cells have already passed clinical phase I trials that confirmed that its infusion is safe [15,16]. Some of the patients with very advanced cancer experienced a major clinical response or a prolonged stable phase. Here we show that NK-92 can be targeted to antigen-expressing lymphoid malignancies by transfecting them with an mRNA-based anti-CD19-CAR. CAR-modified NK-92 cells become capable of effectively killing CD19 positive cell lines as well as primary CLL cells that were resistant to unmodified NK-92. Of note is that transfection efficiency with mRNA was about 10 times higher than with a cDNA plasmid.
Transfection of mRNA, either by physical or viral-based techniques, is preferable to transfection of DNA for several reasons. mRNA is not integrated into the genome and hence does not carry the risk of genomic mutation or instability. mRNA is processed in the cytoplasm and expression occurs immediately after cell membrane penetration, without the need to enter the nucleus. It is also degradable and completely disappears from transfected cells. In addition to these characteristics, we observed that the viability of cells transfected with mRNA was much greater than with DNA, probably because driving mRNA through the cell membrane requires less harsh transfection conditions than necessary for DNA to reach the nucleus. Expression of the protein lasted for 48 h to over 100 h, depending on the protein, which would be sufficient to expect some in vivo interaction with tumor targets. The transfection efficiency in NK-92 cells was >75% with GFP mRNA using a Biorad electroporation equipment, showing that high yields can be easily achieved using a relatively simple and low cost protocol. In contrast, we have not been successful in achieving good yields by electroporating fresh or expanded human NK cells from peripheral or cord blood cells, either with mRNA- or DNA-based vectors (<10% expression, unpublished data). Transfection of DNA and mRNA into several immune type cells was successfully reported by Rabinovich et al. [25], using the Amaxa Nucleofector system (Amaxa Biosystems, Cologne, Germany) with tailored transfection conditions. However, this system did not improve transfection efficiency in our experiments, probably due to some intrinsic properties of NK cells. Peripheral blood or cord blood NK cells may be more amenable to non-physical transfection modalities, such as lentivirus-based constructs, in order to achieve good expression yields [26].
Importantly, NK-92 cells did not lose cytolytic activity after irradiation with 10 Gy, which is the dose required by the FDA when NK-92 are to be infused into patients [15]. Cytotoxicity was most efficient when cells were irradiated immediately before the assay, that is 20 h after electroporation when protein expression is peaking, which would be the time to administer the transfected NK-92. The safety of NK-92 infusions have already been shown in phase I trials but unmodified NK-92 cells do not kill primary CLL cells. Since large-scale GMP compliant electroporation systems (Max-Cyte) are available, CAR-expressing NK-92 could enter treatment protocols.
Acknowledgments
We thank Dr. James Weitzman and Dr. Arthur Rabinovitz for providing us with blood samples from CLL patients.
Footnotes
Conflict of interest statement
H. Klingemann is co-founder and shareholder in ZelleRx Corp.
References
- 1.Shanafelt TD, Byrd JC, Call TG, Zent CS, Kay NE. Narrative review: initial management of newly diagnosed, early-stage chronic lymphocytic leukemia. Ann Intern Med. 2006;145(6):435–47. doi: 10.7326/0003-4819-145-6-200609190-00007. [DOI] [PubMed] [Google Scholar]
- 2.Rozman C, Bosch F, Montserrat E. Chronic lymphocytic leukemia: a changing natural history? Leukemia. 1997;11(6):775–8. doi: 10.1038/sj.leu.2400679. [DOI] [PubMed] [Google Scholar]
- 3.Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol. 2008;9(5):495–502. doi: 10.1038/ni1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. 2008;9(5):503–10. doi: 10.1038/ni1582. [DOI] [PubMed] [Google Scholar]
- 5.Farag SS, Fehniger TA, Ruggeri L, Velardi A, Caligiuri MA. Natural killer cell receptors: new biology and insights into the graft-versus-leukemia effect. Blood. 2002;100(6):1935–47. doi: 10.1182/blood-2002-02-0350. [DOI] [PubMed] [Google Scholar]
- 6.Moretta L. NK cell-mediated immune response against cancer. Surg Oncol. 2007;16(Suppl 1):S3–5. doi: 10.1016/j.suronc.2007.10.043. [DOI] [PubMed] [Google Scholar]
- 7.Verheyden S, Demanet C. NK cell receptors and their ligands in leukemia. Leukemia. 2008;22(2):249–57. doi: 10.1038/sj.leu.2405040. [DOI] [PubMed] [Google Scholar]
- 8.Eshhar Z. The T-body approach: redirecting T cells with antibody specificity. Handb Exp Pharmacol. 2008;181:329–42. doi: 10.1007/978-3-540-73259-4_14. [DOI] [PubMed] [Google Scholar]
- 9.Cooper LJ, Ausubel L, Gutierrez M, Stephan S, Shakeley R, Olivares S, et al. Manufacturing of gene-modified cytotoxic T lymphocytes for autologous cellular therapy for lymphoma. Cytotherapy. 2006;8(2):105–17. doi: 10.1080/14653240600620176. [DOI] [PubMed] [Google Scholar]
- 10.Uherek C, Tonn T, Uherek B, Becker S, Schnierle B, Klingemann HG, et al. Retargeting of natural killer-cell cytolytic activity to ErbB2-expressing cancer cells results in efficient and selective tumor cell destruction. Blood. 2002;100(4):1265–73. [PubMed] [Google Scholar]
- 11.Muller T, Uherek C, Maki G, Chow KU, Schimpf A, Klingemann HG, et al. Expression of a CD20-specific chimeric antigen receptor enhances cytotoxic activity of NK cells and overcomes NK-resistance of lymphoma and leukemia cells. Cancer Immunol Immunother. 2008;57(3):411–23. doi: 10.1007/s00262-007-0383-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gong JH, Maki G, Klingemann HG. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia. 1994;8(4):652–8. [PubMed] [Google Scholar]
- 13.Reid GS, Bharya S, Klingemann HG, Schultz KR. Differential killing of pre-B acute lymphoblastic leukaemia cells by activated NK cells and the NK-92 ci cell line. Clin Exp Immunol. 2002;129(2):265–71. doi: 10.1046/j.1365-2249.2002.01919.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Romanski A, Uherek C, Bug G, Muller T, Rossig C, Kampfmann M, et al. Re-targeting of an NK cell line (NK92) with specificity for CD19 efficiently kills human B-precursor leukemia cells. ASH Annual Meeting Abstracts. 2004;104(11):2747. [Google Scholar]
- 15.Arai S, Meagher R, Swearingen M, Myint H, Rich E, Martinson J, et al. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma—a phase I trial. Cytotherapy. 2008;10(6):625–32. doi: 10.1080/14653240802301872. [DOI] [PubMed] [Google Scholar]
- 16.Tonn T, Becker S, Esser R, Schwabe D, Seifried E. Cellular immunotherapy of malignancies using the clonal natural killer cell line NK-92. J Hematother Stem Cell Res. 2001;10(4):535–44. doi: 10.1089/15258160152509145. [DOI] [PubMed] [Google Scholar]
- 17.Klingemann HG, Gong HJ, Maki G, Horsman DE, Dalal BI, Phillips GL. Establishment and characterization of a human leukemic cell line (SR-91) with features suggestive of early hematopoietic progenitor cell origin. Leuk Lymphoma. 1994;12(5–6):463–70. doi: 10.3109/10428199409073789. [DOI] [PubMed] [Google Scholar]
- 18.Boissel L, Tuncer HH, Betancur M, Wolfberg A, Klingemann H. Umbilical cord mesenchymal stem cells increase expansion of cord blood natural killer cells. Biol Blood Marrow Transplant. 2008;14(9):1031–8. doi: 10.1016/j.bbmt.2008.06.016. [DOI] [PubMed] [Google Scholar]
- 19.Klingemann HG, Wong E, Maki G. A cytotoxic NK-cell line (NK-92) for ex vivo purging of leukemia from blood. Biol Blood Marrow Transplant. 1996;2(2):68–75. [PubMed] [Google Scholar]
- 20.Rai KR, Peterson BL, Appelbaum FR, Kolitz J, Elias L, Shepherd L, et al. Fludarabine compared with chlorambucil as primary therapy for chronic lymphocytic leukemia. N Engl J Med. 2000;343(24):1750–7. doi: 10.1056/NEJM200012143432402. [DOI] [PubMed] [Google Scholar]
- 21.Byrd JC, Rai K, Peterson BL, Appelbaum FR, Morrison VA, Kolitz JE, et al. Addition of rituximab to fludarabine may prolong progression-free survival and overall survival in patients with previously untreated chronic lymphocytic leukemia: an updated retrospective comparative analysis of CALGB 9712 and CALGB 9011. Blood. 2005;105(1):49–53. doi: 10.1182/blood-2004-03-0796. [DOI] [PubMed] [Google Scholar]
- 22.Hillmen P, Skotnicki AB, Robak T, Jaksic B, Dmoszynska A, Wu J, et al. Alemtuzumab compared with chlorambucil as first-line therapy for chronic lymphocytic leukemia. J Clin Oncol. 2007;25(35):5616–23. doi: 10.1200/JCO.2007.12.9098. [DOI] [PubMed] [Google Scholar]
- 23.Fujita Y, Rooney CM, Heslop HE. Adoptive cellular immunotherapy for viral diseases. Bone Marrow Transplant. 2008;41(2):193–8. doi: 10.1038/sj.bmt.1705906. [DOI] [PubMed] [Google Scholar]
- 24.Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. 2005;105(8):3051–7. doi: 10.1182/blood-2004-07-2974. [DOI] [PubMed] [Google Scholar]
- 25.Rabinovich PM, Komarovskaya ME, Ye ZJ, Imai C, Campana D, Bahceci E, et al. Synthetic messenger RNA as a tool for gene therapy. Hum Gene Ther. 2006;17(10):1027–35. doi: 10.1089/hum.2006.17.1027. [DOI] [PubMed] [Google Scholar]
- 26.Su S, Muthalagu R, Nguyen D, Smith A, Keyvanfar K, Sheila Rao S, et al. Effective and stable gene transfer into human NK cells using an HIV-1-based lentiviral vector system. American Society of Gene Therapy 11th Annual Meeting; 2008. [Google Scholar]