

Abstract
Advances in the design of potential bone-selective drugs for the treatment of various bone-related diseases are creating exciting new directions for multiple unmet medical needs. For bone-related cancers, off-target/non-bone toxicities with current drugs represent a significant barrier to the quality of life of affected patients. For bone infections and osteomyelitis, bacterial biofilms on infected bones limit the efficacy of antibiotics because it is hard to access the bacteria with current approaches. Promising new experimental approaches to therapy, based on bone-targeting of drugs, have been used in animal models of these conditions and demonstrate improved efficacy and safety. The success of these drug-design strategies bodes well for the development of therapies with improved efficacy for the treatment of diseases affecting the skeleton.
Keywords: Bone targeting, multiple myeloma, osteomyelitis, biofilm, bisphosphonate, antibiotic, conjugate, bone resorption
INTRODUCTION
The ultimate goal of drug design is to develop therapies that work directly on the tissues, cells, and biochemical targets relevant to any specific disease, and do not affect the biochemistry at any other non-diseased compartment of the body. Calcified tissues of the skeleton are targeted with great specificity by the bisphosphonate drug class to treat bone-related diseases with minimal side effects on soft tissues. This has been evident for many years with the potent nitrogen-containing bisphosphonates for the treatment of bone diseases, including osteoporosis and cancers that have metastasized to bone (Pazianas, Cooper, Ebetino & Russell, 2010). Bisphosphonates are a group of compounds with the general structure: (HO)2P(O)CR1R2P(O)(OH)2 (Figure 1A). Their strong bone-binding affinity is predominantly determined by the two phosphonate groups (P-C-P), which form strong bi- and tri-dentate interactions with calcium, to which the R1 and R2 groups on the bridging carbon also contribute to a certain extent (Ebetino et al., 2011; Russell, Watts, Ebetino & Rogers, 2008). Delivery of other drug classes specifically to bone by linking them to bisphosphonates is an intriguing therapeutic approach that has not yet found clinical utility (Cole, Vargo-Gogola & Roeder, 2016; Farrell, Karpeisky, Thamm & Zinnen, 2018; Kempfle et al., 2018; Young & Grynpas, 2018). Although this topic has been covered recently in general reviews (Cole, Vargo-Gogola & Roeder, 2016; Farrell, Karpeisky, Thamm & Zinnen, 2018; McKenna, Haratipour, Duro & Ebetino, 2020; Xing et al., 2020), in this paper we assess the success of this approach to two areas of bone disease that continue to be unmet medical needs. Specifically, we review work in which bisphosphonates are chemically linked to drugs used clinically to treat multiple myeloma (Wang, Xiao et al., 2018; Wang et al., 2020) or bone infections (osteomyelitis) (Sedghizadeh et al., 2017b) that have been used successfully as drug-releasing conjugates in animal models of these diseases.
FIGURE 1.

(a)“Target-and-release”design of bisphosphonate-drug conjugates. (b) Structures of bisphosphonate conjugates of bortezomib (BP-Btz), chloroquine (BP-CQ), and hydroxychloroquine (BP-HCQ). (c–h) A combination of BP-Btz and BP-CQ kills more Btz-resistant U266 human myeloma cells or primary myeloma cells from patients with relapsed/refractory MM than a single agent. (c–f) Human U266 cells were treated with various drugs for 72 h. Cell survival was determined using a CCK8 kit. The percentage of survival was calculated using Veh-treated cells as 100%. Btz-sensitive/parental U266 cells (U266.P) and Btz-resistant U266 cells (U266.BR) were treated with different doses of Btz (c) or BP-Btz (d). U266. BR cells were treated with 6 nM Btz, 30 μM CQ, Btz + CQ (e) or 30 μM BP, 6 nM BP-Btz, 30 μM BP-CQ, BP-Btz + BP-CQ (f). n = 2 repeats with similar results. Bone marrow aspirates of Patients #1–3 with relapsed/refractory MM (g) and Patients #4–5 with newly diagnosed MM (h) were used. CD138+ cells were isolated with anti-CD138 antibody-conjugated magnetic beads via positive selection. Cells were treated with various drugs for 24 h. The doses for cells from Patients #1, 2, and 4 were the same as in (f), and the doses for patient #3 were 12 nM BP-Btz and 40 μM BP-CQ, and for patient #5 were 6 nM BP-Btz, 20 μM BP-CQ, or BP-HCQ. *P<0.05, significantly different as indicated. Figure adapted from Boyce, 2019; Tao et al., 2018 and Xing et al., 2020.
The ability of bisphosphonate-based pharmaceuticals to detect bone cancers using skeletal scintigraphy has been recognized for many years. Technetium-labeled bisphosphonate complexes have been used in clinical bone scanning for decades (Cole, Vargo-Gogola & Roeder, 2016). Recent developments in the targeting of a nucleoside monophosphate with etidronate have led to a clinical candidate for bone cancers (Zinnen, Karpeisky, Von Hoff, Plekhova & Alexandrov, 2019). Our team at the University of Rochester has also demonstrated enhanced efficacy and evidence of better off-target safety by combining bortezomib with a pharmacologically inactive bisphosphonate for the treatment of multiple myeloma in a mouse model (Wang, Cai, et al., 2018). Thus, bisphosphonate-based bone targeting to create potent and selective therapies to treat bone neoplasms and other bone diseases is receiving increasing attention. The challenges inherent in combining two drugs into one molecule are also being identified and addressed. For example, it is clear that the use of relatively inert bisphosphonates for this purpose reduces the chance of confounding results in vivo, particularly on bone resorption. Additionally, an ideal linkage between the bisphosphonate and the drug “warhead” being delivered to the bone surface should have a slow rate of cleavage in plasma to allow delivery to the bone compartment, but must then be labile once adsorbed to the bone surface to allow sufficiently rapid release of the drug to achieve a therapeutically effective local concentration (Figure 1A). Certain properties of the microenvironment in bone and osteoclast resorption lacunae, e.g., the generally acidic pH environment, and some specific enzymes secreted by osteoclasts, the cells that degrade bone, make selective cleavable linkage design possible.
1. Changes in Normal Bone Remodeling
The bone remodeling process is regulated by two major cells: osteoclasts mediate bone resorption, while osteoblasts form bone. In a normal skeleton, it has been shown by Fogelman and others with bisphosphonate-based technetium scanning that there is a greater % of bone remodeling in the trabecular regions of bone and therefore greater uptake of bisphosphonate because of the higher percentage of exposed mineral surface and mineralizing osteoid matrix to which bisphosphonates bind (Fogelman, 1982). The bisphosphonates coordinate and chemisorb exquisitely well with the calcium ions of hydroxyapatite surfaces and either bone resorption or formation events lead to exposed bone mineral. Higher uptake of bisphosphonates on the skeleton is therefore associated with bone turnover. Abnormal bone remodeling is characterized by either an imbalance of the resorption and formation cycle and/or by a more rampant rate of either or both processes. In many diseases, such as Paget’s disease, or in the presence of osteomyelitis or bone metastases, this excessive rate of remodeling can be observed focally at the site of lesions. Thus, more remodeling leads to greater exposure of bone mineral (hydroxyapatite surface area) and therefore greater uptake of bisphosphonates focally at diseased skeletal sites.
Multiple Myeloma
Multiple myeloma (MM) is a hematologic malignancy caused by the expansion of malignant plasma cells within the bone marrow (BM), thereby crowding out the normal marrow cells and leading to anemia and other cytopenias, and to high levels of monoclonal proteins in serum and urine (Al-Farsi, 2013; El Arfani, De Veirman, Maes, De Bruyne & Menu, 2018; Shain, Dalton & Tao, 2015). Myeloma cells are highly dependent on the BM microenvironment for growth, survival and activation through interactions particularly with BM stromal cells (Xu, De Veirman, De Becker, Vanderkerken & Van Riet, 2018), osteoblasts (Kawano et al., 2015) and osteoclasts (Lawson et al., 2015). However, myeloma cells also damage the BM microenvironment (Ghobrial, Detappe, Anderson & Steensma, 2018) involving multiple cell types (Al-Farsi, 2013; Mahindra, Hideshima & Anderson, 2010). In particular, MM cells inhibit osteoblastogenesis by secreting soluble factors, such as Dkk1, sFRP2 and sclerostin, and suppressing Runx-Related Transcription Factor 2 (Runx2) activity through direct cell-to-cell contact with the involvement of VLA-4/VCAM-1 interaction. Concurrently, MM cells promote osteoclast function by releasing numerous cytokines, such as RANKL (Giuliani & Rizzoli, 2007; Terpos, Ntanasis-Stathopoulos, Gavriatopoulou & Dimopoulos, 2018). Thus, myeloma cells increase bone resorption and decrease bone formation, inducing a severe imbalance in bone remodeling and contributing to the development of myeloma-associated bone diseases, including focal lytic lesions, pathological fractures, and hypercalcemia (Paton-Hough, Chantry & Lawson, 2015). Although recent advances in treatment have greatly improved patient outcomes, MM remains largely incurable (Goldschmidt, Ashcroft, Szabo & Garderet, 2019).
1.1. Bortezomib
Bortezomib (Btz, Velcade®) is a drug used to treat patients with MM and mantle cell lymphoma (Jin et al., 2018; Manasanch & Orlowski, 2017; Robak et al., 2015), and its ability to treat other types of cancer is being tested in clinical trials (clinicaltrials.gov, 2019). Btz is a very specific 26S proteasome inhibitor, which reversibly but strongly binds the threonine proteases of the 20S subunit, enabling inhibition of the hydrolyzing catalytic activities of the 26S proteasome (Painuly & Kumar, 2013). Mechanistically, Btz directly inhibits proteasomal degradation of the large amounts of immunoglobulin produced by myeloma cells. This allows the accumulation of these useless/ineffective proteins, leading to endoplasmic reticulum stress, activation of the unfolded protein response, and eventually to myeloma cell apoptosis (Brewer & Diehl, 2000; Manasanch & Orlowski, 2017; Obeng, Carlson, Gutman, Harrington, Lee & Boise, 2006; Zinszner et al., 1998). Btz may also inhibit NF-κB signaling by protecting IκBα from degradation, thus preventing its pro-survival activity and promoting myeloma cell apoptosis (Roy, Sarkar & Basak, 2018; Vrabel, Pour & Sevcikova, 2019). However, other studies have reported that instead of inhibiting IκBα degradation, Btz can decrease the expression of IκBα, resulting in NF-κB activation in various tumor cell lines, including MM (Hideshima et al., 2009; Li et al., 2010). Btz also inhibits osteoclast formation by modulating p38, activator protein-1, and NF-κB signaling pathways (von Metzler et al., 2007; Zavrski et al., 2005), and promotes osteoblast differentiation by stabilizing the key osteoblast transcription factor, Runx2. Therefore, Btz also suppresses bone resorption and stimulates bone formation, which can ameliorate the abnormal bone homeostasis caused by MM (Giuliani et al., 2007; Pennisi, Li, Ling, Khan, Zangari & Yaccoby, 2009). These complementary functions of Btz on myeloma cells and bone cells place it as an ideal drug for treating patients with MM. Btz is administrated intravenously or subcutaneously. Similar to other anti-cancer drugs given by systemic administration, the off-target effects of Btz on non-skeletal tissues, mainly peripheral neuropathy (Cavaletti et al., 2007) and thrombocytopenia (Lonial et al., 2005; Murai et al., 2014; Shi et al., 2014), have restricted the amounts of the drug that can be administered. More importantly, patients develop inherent or acquired Btz resistance (BR), which limits its clinical efficacy (Dispenzieri, Jacobus, Vesole, Callandar, Fonseca & Greipp, 2010; Kuhn et al., 2012; Murray, Auger & Bowles, 2014; Richardson et al., 2003; Richardson et al., 2005; Ruschak, Slassi, Kay & Schimmer, 2011). The development of BR involves multiple mechanisms, such as activation of chemo-resistance pathways, immunoproteasomes, and mutations in proteasome subunits (Niewerth, Jansen, Assaraf, Zweegman, Kaspers & Cloos, 2015). Agents targeting these molecular pathways have been tested in preclinical studies using BR myeloma cell lines and in a set of CD138+ cells obtained from BM of patients with relapsed/refractory myeloma (Chauhan et al., 2010; Richardson et al., 2014; Zangari & Suva, 2016).
1.2. Autophagy and its inhibitors, Chloroquine and Hydroxychloroquine, in treatment of multiple myeloma
Recent studies report that autophagy may play a role in drug resistance in cancer patients (Sui et al., 2013) including MM (Abdel Malek et al., 2015; Hamouda et al., 2014; Zhang et al., 2015). Autophagy is a lysosome-dependent protein degradation process. Autophagy degrades unnecessary or dysfunctional cellular components and releases nucleotides, amino acids or fatty acids from them (Yun et al., 2017) as a source of energy to ensure cell survival under critical conditions, such as hypoxia and starvation (Verbaanderd et al., 2017). However, extensive autophagy may also lead to cell death (Tsujimoto & Shimizu, 2005). In myeloma cells, massive amounts of immunoglobulin that are not degraded because of Btz-mediated proteasome inhibition are proposed to serve as an energy source via autophagy, thereby exerting a pro-survival effect under drug treatment (Attar-Schneider, Drucker, Zismanov, Tartakover-Matalon, Rashid & Lishner, 2012; Auner & Cenci, 2015; Dykstra, Allen, Born, Tong & Holstein, 2015; Hoang, Benavides, Shi, Frost & Lichtenstein, 2009; Zeng, Chen, Zhao & Cui, 2012). Thus, inhibition of autophagy may kill drug-resistant myeloma cells by limiting energy supply.
Chloroquine (CQ) and Hydroxychloroquine (HCQ) are FDA-approved autophagy inhibitors and have been used to treat patients with malarial infections (CQ), systemic lupus erythematosus and rheumatoid arthritis (HCQ) for decades. Recent reports indicate that both CQ and HCQ reduce bone resorption by inhibiting osteoclast formation (Both et al., 2018; Xiu et al., 2014). They have been extensively studied both in vitro and in vivo in various cancer types. Results show that CQ or HCQ effectively increase the efficacy and limit drug resistance of standard anti-cancer therapies (Verbaanderd et al., 2017). Recently, 4 clinical studies (ClinicalTrials.gov Identifier: NCT01438177; NCT00568880; NCT01396200; NCT01689987 (Verbaanderd et al., 2017)) have investigated the use of CQ or HCQ in combination with other anti-MM drugs in patients with relapsed/refractory myeloma. A phase I clinical trial (NCT00568880) led by Dr. Dan Vogl (Vogl et al., 2014) and a phase II trial (NCT01438177) led by Dr. Amitabha Mazumder (Montanari, Lu, Marcus, Saran, Malankar & Mazumder, 2014) examined the effects of a combination of HCQ and Btz in 25 cases with refractory or relapsed MM, or CQ, Btz, and Cyclophosphamide in 11 similar patients. Both trials report promising results and demonstrated that CQ or HCQ was able to partially restore Btz sensitivity (Montanari, Lu, Marcus, Saran, Malankar & Mazumder, 2014; Vogl et al., 2014). However, some serious adverse events, including hypoglycemia (Goyal & Bordia, 1995; Prabha, 1996), BM suppression (Nagaratnam, Chetiyawardana & Rajiyah, 1978), cardiomyopathy (Costedoat-Chalumeau et al., 2007) and irreversible retinal toxicity (Browning, 2014) have been associated with long-term treatment with CQ or HCQ. Some subjects suffered from the toxic side effects of the combined regimen (Montanari, Lu, Marcus, Saran, Malankar & Mazumder, 2014) that led some of them to stop therapy (Vogl et al., 2014). Low doses of CQ or HCQ in combination with Btz and/or other drugs limited by the systemic adverse effects have been proposed to be responsible for hindering achievement of more robust clinical responses (Scott et al., 2017; Vogl et al., 2014).
1.3. Preclinical studies using bone-targeted strategies to deliver Bortezomib or other drugs to bone marrow in mouse models of MM
Given that MM primarily originates in the BM and destroys bone, selective delivery of effective anticancer drugs such as Btz specifically to bone has been a focus of myeloma research. Bone targeting should reduce the toxic side effects arising from systemic distribution of Btz and make dose escalation possible in order to improve therapeutic outcomes. In 2010, Reinholz et al. linked the chemotherapeutic drug arabinocytidine-5′-phosphate to a bisphosphonate (BP) drug, etidronate, and generated a series of bone-targeted anti-cancer compounds, of which the leading candidate was named MBC-11. MBC-11 was designed to target both osteoclasts (etidronate) and tumor cells (arabinocytidine-5′-phosphate), and it reduced bone metastases and bone tumor burden in mice-bearing 4T1/luc mouse breast cancer cells following mammary gland inoculation. It inhibited bone destruction and increased overall survival in mice bearing KAS-6/1-MIP1α human myeloma cells following tail vein injection (Reinholz et al., 2010). Subsequently, MBC-11 advanced to a phase I oncology clinical trial (NCT02673060) in 2014 for patients with cancer-induced bone disease and achieved partial efficacy (Zinnen, Rowinsky, Alexandrov, Plekhova, Roudas & Karpeisky, 2017; Zinnen, Karpeisky, Von Hoff, Plekhova & Alexandrov, 2019). In 2013, Agyin et al. synthesized a series of BP (alendronate)-proteasome inhibitor conjugates and showed that they had similar effects as their non-bone-targeted counterparts to kill MM cells in vitro (Agyin, Santhamma & Roy, 2013). In 2014, Swami et al. engineered bone-homing polymeric nanoparticles (NPs), BP (alendronate)-conjugated polymer PLGA-b-PEG-Ald, to load Btz and demonstrated that the bone-targeted NPs could bind to bone fragments in vitro and retain and accumulate in bone in vivo. Pretreatment of Btz loaded into bone-targeted NPs enhanced survival and decreased tumor burden in mice bearing Luc+/GFP+ MM1S human myeloma cells (Swami et al., 2014). In 2018, 2 studies reported the synthesis of bone-targeted nanoparticles (NPs), loaded Btz with a pH-sensitive aryl boric acid ester linkage that offers a pH-responsive drug release mechanism (Wang, Cai, et al., 2018; Zhu et al., 2018). In both studies, in vitro release assays showed that only 20-30% of Btz was released from the conjugated drugs in neutral buffer (pH 7.4), while under acidic conditions of around pH 5.0, the release rate of Btz was faster, delivering as much as 60-70% of the drug load. In mice with intra-tibial administration of MDA-MB-231 cells, bone-targeted Btz significantly reduced tumor burden and bone destruction relative to free Btz-treated mice.
Although some advances were achieved by using the above bone-targeted agents to treat MM or metastatic bone tumors, the possibly reduced systemic adverse effects of these drugs, especially the Btz derived compositions, have not been formally investigated. Furthermore, most studies have used an antiresorptive BP, such as etidronate (Reinholz et al., 2010; Zinnen, Rowinsky, Alexandrov, Plekhova, Roudas & Karpeisky, 2017) or alendronate (Agyin, Santhamma & Roy, 2013; Swami et al., 2014; Zhu et al., 2018). With this approach, it can be difficult to distinguish the pharmacological effect of the BP component from that of released Btz. Recently, we linked Btz to a bisphosphonate (BP) (Figure 1B) that binds avidly to bone, but is not antiresorptive, using a novel covalently bonded linker to generate a BP-linked Btz (named BP-Btz) conjugate and demonstrated that BP-Btz, but not Btz, bound to bone slices and inhibited the growth of myeloma cells in vitro. BP-Btz more effectively reduced tumor burden and bone loss than Btz in the 5TGM1 mouse model of MM (Wang, Xiao et al., 2018). In particular, we also demonstrated that BP-Btz generated significantly less systemic adverse effects, such as thrombocytopenia (Wang, Xiao, et al., 2018) and peripheral neuropathy (Wang et al., 2020), compared with Btz alone.
BPs bind preferentially, but not solely, to skeletal sites with high bone turnover (Russell, Watts, Ebetino & Rogers, 2008). Thus, some BP-conjugates which might be delivered to other skeletal sites could cause bone side effects. For example, BP-conjugates that utilize BPs with significant antiresorptive function (Agyin, Santhamma & Roy, 2013; Reinholz et al., 2010; Zinnen, Karpeisky, Von Hoff, Plekhova & Alexandrov, 2019) could in theory exhibit adverse effects that have been associated with other bisphosphonates, including osteonecrosis of the jaw, severe suppression of bone turnover, and atypical subtrochanteric femoral fractures (Abrahamsen, 2010; Keller, 2014). Therefore, in our conjugate approach, we designed BP moieties, which could bind avidly to the bone, but lack significant antiresorptive activity, to limit any chance of side effects and to clarify the source of pharmacological effect in our studies. Furthermore, even the effects of delivery to lower bone turnover skeletal sites would likely be relatively minimal, since less turnover involving osteoclast activity would likely lead to a slower release of the active warhead at those sites. Regarding the free drug, Btz promotes osteoblast differentiation, and reduces osteoclast formation (Giuliani et al., 2007; Uy et al., 2007), while chloroquine (Xiu et al., 2014) or hydroxychloroquine (Both et al., 2018) reduces osteoclastogenesis, which are features that may be beneficial to MM therapy, since a drastic loss of bone is observed in patients with this cancer. Although 99m-technetium bone scans indicate a high preference for bisphosphonates to localize at sites of higher turnover where their primary effects are likely to occur, we do not currently have data to indicate how much BP or free drug will be delivered to bone sites with lower bone turnover and what side effects this might lead to. Isotope labeling and related pharmacokinetic and pharmacodynamic studies will help to evaluate these important questions.
1.4. Effects of BP-Btz and BP-CQ or BP-HCQ on Bortezomib-resistant MM
As mentioned above, previous studies suggest that Btz resistance might be attributed to autophagy (Abdel Malek et al., 2015; Hamouda et al., 2014; Zhang et al., 2015), and the autophagy inhibitors, CQ or HCQ, were able to improve sensitivity to Btz or other anti-MM drugs to relapsed/refractory MM in clinical studies (Montanari, Lu, Marcus, Saran, Malankar & Mazumder, 2014; Vogl et al., 2014). However, the efficacy was limited due to several systemic adverse effects, hindering the use of higher doses of CQ or HCQ or Btz (Vogl et al., 2014). In view of the findings that targeting Btz to bone successfully improved its efficacy and ameliorated off-target adverse effects (Wang, Xiao et al., 2018), we also generated a bone-targeted CQ (BP-CQ) and a bone-targeted HCQ (BP-HCQ) by conjugating them to a bisphosphonate using linker chemistry similar to that for BP-Btz (Figure 1B) (Boeckman, Boyce, Xiao, YAO & Ebetino, 2017; Xing et al., 2020; Yao et al., 2016). Our rationale is that delivering Bortezomib or chloroquine conjugates to bone using a BP with high bone affinity, but no significant antiresorptive effects, could result in high and persistent local drug concentrations at sites of bone diseases, thereby killing more myeloma cells and preventing bone loss, with fewer adverse effects.
In our initial study, we examined if BP-Btz plus BP-CQ or BP-HCQ could more effectively kill BR myeloma cells than individual drug alone in vitro. We used a BR U266 human myeloma cell line (Mitra et al., 2016) and found that they were resistant to both Btz and BP-Btz to a similar extent (Figure 1C&D). Treatment of the BR U266 human myeloma cell line with 6 nM Btz or 30 μM CQ for 72 hrs killed 40% of the cells, while Btz+CQ killed 70%, and BP-Btz and BP-CQ had similar effects in vitro (Figure 1E&F). We also demonstrated that the equimolar concentration of the BP, including the linker structure that is likely to be generated on release of Btz, had no greater effect on myeloma cells than on vehicle-treated cells (Figure 1F). We then tested the effects of BP-Btz plus BP-CQ on primary myeloma cells that were isolated from BM of 3 patients with relapsed/refractory MM (Figure 1G). We used the same doses of Btz (6 nM) and CQ (30 μM) for cells from patient #1 and #2 and found that BP-Btz or BP-CQ alone killed 20-30% of myeloma cells, while a combination of these killed 50%. To determine if increasing the dose of Btz or CQ could achieve a better effect, we treated cells from patient #3 with high doses of Btz (12 nM) and CQ (40 μM) and found that they had a similar killing effect as the lower doses. We also examined the effects of BP-Btz and BP-CQ on CD138+ myeloma cells from 2 newly diagnosed MM patients. Cells from one patient responded well to Btz, with 6 nM killing 65%, while 30 μM BP-CQ killed 70%, and the combination killed 90% of the patient’s myeloma cells (Figure 1H). However, cells from another patient responded poorly to BP-Btz, with 6 nM BP-Btz killing 15%, while 20 μM BP-CQ or BP-HCQ killed 25%, and the combination killed 50% of the patient’s myeloma cells (Figure 1H), suggesting that myeloma cells from the second case may already be Btz-resistant. These data that have been presented in several international conferences (Boyce, 2019; Tao et al., 2018) and discussed in our recent review paper (Xing et al., 2020) suggest that combined BP-Btz and BP-CQ or BP-HCQ therapy may overcome BR myeloma and represent a new treatment for relapsed/refractory MM patients or newly diagnosed MM patients who are inherently BR.
Studies in the literature have estimated the local pH under the tight sealing zone of the active osteoclast on the bone surface. Mechanistically, it is accepted that the pH in the resorption lacunae is acidic to account for rapid dissolution of bone mineral mediated by the cell. Silver et al (Silver, Murrills & Etherington, 1988) reported that the local pH under the active osteoclast sealing zone is as low as 4.5. A more recent paper reported a pH range of 4-6 in the resorption pit (Kowada et al., 2011). Thus, we designed the linkage in BP-Btz to be acid sensitive. The in vitro experiments were performed at physiological pH (around 7.2), and we did not observe a significant difference between the effect of free drug and bone-targeted drug on inhibition of MM cell growth. Since free drug appears to be released primarily at an acidic pH, we speculate that the free drug is released under the osteoclast or within lysosomes or other cell organelles where the pH is low after the entire conjugate enters the cell. These release mechanisms could be explored further using fluorescent- or isotope-labeled BPs and BP-Btz.
1.5. Conclusion
Despite current clinical treatments for MM, including Btz, showing remarkable efficacy, drug resistance and off-target effects have restricted the use of Btz clinically. Several clinical studies have also shown some promising results of the autophagy inhibitors, CQ and HCQ, to overcome Btz resistance. However, the side effects of CQ or HCQ limit the use of more effective doses, hindering achievement of more robust clinical responses. Development of novel bone-targeted therapeutic agents in recent years has significantly improved efficacy in preclinical studies in the treatment of MM. Although some critical information, such as drug release kinetics, possible synergies of BP-drug payload interactions, mechanisms of action, tissue distribution, and toxicity, still need to be gathered, the BP conjugates represent a promising therapeutic strategy.
2. Osteomyelitis
Osteomyelitis is a limb- and life-threatening infection of bone that is difficult to treat clinically (Lew & Waldvogel, 2004). These infections are associated with fractures (1.8-27%), with open fractures of the leg produced by high energy impact injury being most common (incidence 27%) (Chen & Vallier, 2016; Pollak, Jones, Castillo, Bosse, MacKenzie & Group, 2010). Other significant sources of these infections are joint replacement surgery (arthroplasty) and secondary infection of the bone after cutaneous infections, which occur for example in diabetic patients. As an example of the burden of this disease, the incidence of osteomyelitis joint infection following total knee or hip replacement is 1.0-3.0% and 0.3-2.4% respectively (Kamath et al., 2015). Among patients who required knee joint replacement or joint prosthesis revision, the primary reason was infection (Bozic et al., 2015). Osteomyelitis is also a major reason for hip joint revision (Kamath et al., 2015). When infection was the reason for joint revision surgery, the associated mortality was reported to be 18% (Choi & Bedair, 2014; Choi, Beecher & Bedair, 2013).
2.1. Current antimicrobial treatment for osteomyelitis
A number of Gram-positive and Gram-negative bacteria, as well as fungi and mycobacteria, can cause osteomyelitis, but by far the most common organism implicated in bone and joint infections is Staphylococcus aureus (SA), both methicillin-susceptible (MSSA) and methicillin–resistant (MRSA) (Dubost et al., 2014; Ferrand et al., 2016). For jawbone infections, Aggregatibacter actinomycetemcomitans (Aa) is a key organism (Stoodley et al., 2011). The standard of care for bone and joint infections usually requires systemic administration of antibiotics, typically vancomycin for MRSA and multidrug-resistant MRSA strains, and fluoroquinolones (ciprofloxacin, moxifloxacin etc.) for gram-negative pathogens (Fraimow, 2009). For acute infections, intravenous antibiotics are generally prescribed for 2 – 6 weeks. Prolonged courses of oral antibiotics may follow for chronic infections, or infections associated with retained implanted hardware. Both for acute and chronic infections, these extended courses of therapy can lead to drug-related adverse events in a significant percentage of patients – 15% in one estimate for a cohort treated for infections with MSSA. This is particularly an issue with vancomycin where nephrotoxicity can occur in as many as 43% of patients (Carreno, Kenney & Lomaestro, 2014). In general, these antibiotic treatments have poor bone pharmacokinetics and limited osseous absorption in vivo (Fong, Ledbetter, Vandenbroucke, Simbul & Rahm, 1986; Kim, Kim & Oh, 2014). Systemic toxicity or adverse effects, and resistance, are clinical issues associated with prolonged dosing regimens.
The inadequate efficacy of current antimicrobial treatments for osteomyelitis has been ascribed to the limited access of systemically administered antibiotics to sites where causative bacteria can reside as biofilms on bone surfaces, even surfaces deep within the osteocytic canalicular network (de Mesy Bently et al., 2017). Sedghizadeh et al. recently demonstrated that osteomyelitis pathogens can invade bone, establish chronic biofilms on bone surfaces, and directly destroy and resorb bone without participation of host immunity or osteoclastogenesis (Junka et al., 2017). These observations add to the difficulty in treating osteomyelitis biofilms, and to the urgency of resolving an unmet medical need in this indication. Therefore, any improvement in bone bioavailability of therapeutic antibiotics would be a significant advance in treating osteomyelitis.
Local antibiotic delivery to treat bone and joint infections can be attempted at the time of surgical debridement using antibiotic-impregnated beads or antibiotic-impregnated cement (Uskokovic, 2015). However, currently marketed beads are not bio-absorbable; therefore, a second procedure is required to remove them. They also tend to release antibiotics in an initial burst pattern that quickly depletes the bulk of the drug from the carrier beads, followed by a slow release of lower concentrations that may not be adequate to control infection and may foster development of resistance. While there have been recent advances in the use of bio-stimuli responsive release strategies (Lavrador, Gaspar & Mano, 2018; Lin, Caldwell, Bhaduri, Goel & Agarwal, 2017), these concerns limit the usefulness of this approach in the majority of bone and joint infections (Bozhkova, Novokshonova & Konev, 2015; Uskokovic, 2015). In the realm of prosthetic joint infection (PJI), antibiotic-impregnated cement is used commonly at the time of first debridement of an infected implant to improve control of the infection. A recent study showed that this can be efficacious in the control of the infection in two-stage hip revision surgeries (Staats et al., 2017). Different cement substrates are available and several antibiotics are used in these treatments, with vancomycin being the most common. Variable anti-bacterial loading and cement characteristics can lead to changes in mechanical properties (Slane, Gietman & Squire, 2018) of the cement implant as well as varying anti-microbial elution rates (Boelch et al., 2018). Concerns about prolonged sub-therapeutic antibiotic concentrations and selection of resistant organisms also apply to cement (Corona, Espinal, Rodriguez-Pardo, Pigrau, Larrosa & Flores, 2014; Walker, Baker, Holleyman & Deehan, 2017).
2.2. Bone-targeted antimicrobial therapy for osteomyelitis
BP drugs have a high affinity for bone and preferentially accumulate at sites of active bone disease or biofilm infection, resorption, and remodeling (Cheong et al., 2014; Ebetino et al., 2011). BPs also penetrate the canalicular network to osteocytes and osteocytic lacunae where no blood flow exists and where S. aureus biofilm organisms are known to embed (de Mesy Bentley et al., 2017). In addition, the BP class of drugs has a long track-record of clinical use and a good safety profile relative to most drug classes, and have thus been utilized for pharmacotherapy in several bone diseases, such as osteoporosis, multiple myeloma, metastatic cancer to bone, and Paget’s disease of bone. To exploit this BP affinity for bone, we are investigating a “target and release” chemistry approach for treating bone infections which involves delivery of antibiotics to the hydroxyapatite (HA) mineral of the skeleton via conjugation to BPs, utilizing serum-stable drug-BP linkers that metabolize and release the antibiotic at bone surfaces and within the canalicular network. Here, we have also utilized pharmacologically “inert” BPs for this purpose to avoid confounding antiresorptive effects from the carrier BP.
We recently reported impressive in vivo efficacy in animal studies with a novel bisphosphonate-carbamate-ciprofloxacin (BCC) conjugate, BV600022 (Sedghizadeh et al., 2017b) (Figure 2A). Quantitative determination of the colony forming units (CFU) of bacteria from resected peri-prosthetic osteomyelitis tissue showed that a single dose of 10 mg/kg of conjugate gave ~2 log reduction of CFU or 99% bacterial killing efficacy and nearly an order of magnitude greater activity than ciprofloxacin alone given in multiple doses (10 mg/kg X 3) (Figure 2B). Furthermore, the effects of a single dose of 10 mg/kg of conjugate in vivo were significantly greater (p=0.0005, unpaired t-test) than the untreated control arm. Also, we synthesized and tested a non-cleavable (non-antibiotic releasing) amide-linked BP conjugate (BAC) BV600026 (Figure 2A), which was found to be nearly inactive against osteomyelitis biofilms grown on HA surfaces in vitro (Sedghizadeh et al., 2017b).
FIGURE 2.
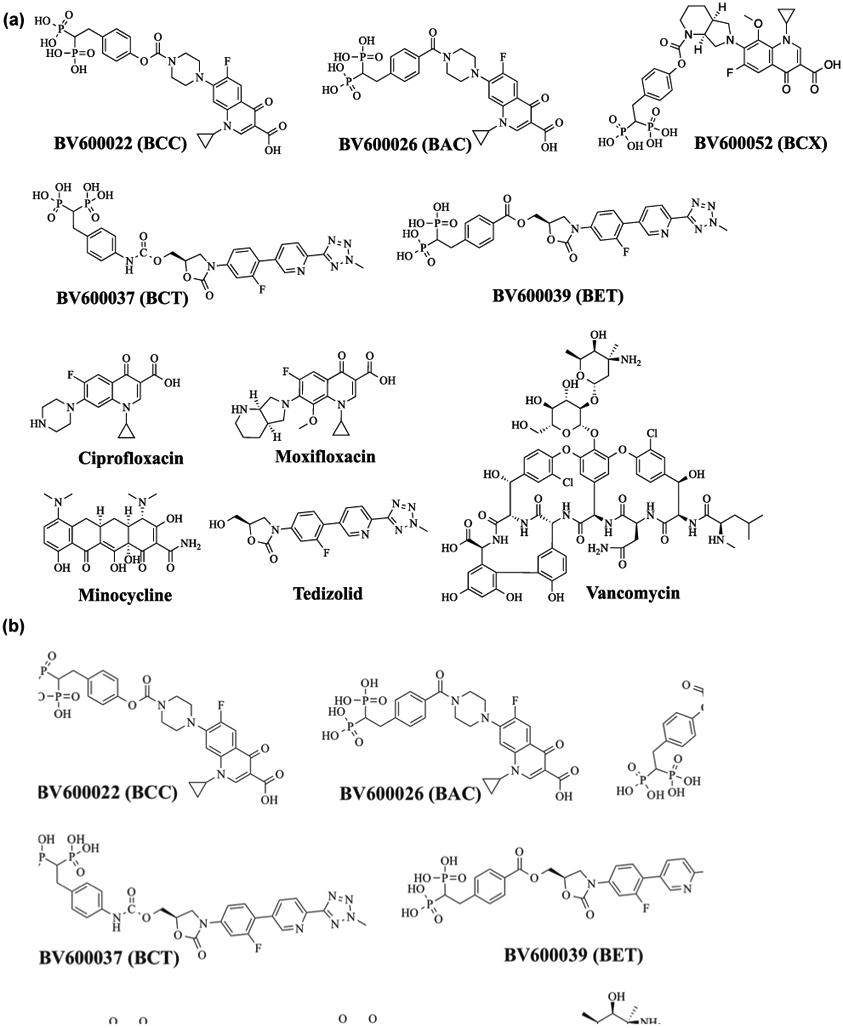
(a) BP-antibiotic conjugates and parent antibiotics used in testing; (b) antimicrobial results in a rat model of periprosthetic osteomyelitis. Data show efficacy of ciprofloxacin and BCC for reducing bacterial load or mean CFU per g tissue (Y-axis). The greatest efficacy was observed at a single high dose (10 mg•kg−1) of BCC where a 2-log reduction (99% bactericidal activity) was seen, compared with the negative control (Figure 2b adapted from Sedghizadeh, Sun, et al., 2017)
Additional in vitro assays of S. aureus and Aa (jawbone) osteomyelitis pathogens in both planktonic and biofilm assays have been performed with BCC treatment as well as with a few other novel BP-antibiotic conjugates (Figure 2A). A new fluoroquinolone carbamate conjugate, bisphosphonate-carbamate-moxifloxacin (BCX, BV600052), based on the later generation and more potent moxifloxacin fluoroquinolone, was compared to BCC in these assays (Sedghizadeh et al., 2017a; Sedghizadeh et al., 2019; Sedghizadeh et al., 2017b; Sedghizadeh et al., 2020). In addition, some of the most important oxazolidinones are currently used against gram-positive pathogens, including superbugs such as MRSA. These antibiotics are considered clinically as choices of last resort when other antibiotics have failed. We therefore also designed, synthesized, and tested two new BP-oxazolidinone conjugates, including a novel BP-carbamate-tedizolid conjugate (BCT, BV600037), and a related more rapidly cleavable BP-ester-tedizolid conjugate (BET, BV600039), for activity against S. aureus. Tedizolid is a recent FDA-approved oxazolidinone antibiotic indicated for the treatment of skin infections, particularly those caused by both methicillin-susceptible S. aureus and methicillin-resistant S. aureus pathogens, which cause the majority of clinical cases of skin infection (Ebetino, Sun, McKenna, Sadrerafi & Cherian, 2020). Since most osteomyelitis cases are also caused by similar Staphylococcal species, the application of tedizolid to bone infections could prove efficacious. An ester-linked conjugate is expected to have more rapid linker release kinetics than a carbamate-linked conjugate, and we projected it could be a good comparison for the identification of the optimal linkage. We also tested various unconjugated antibiotics, including vancomycin (V) and minocycline (M), which are used clinically in the setting of infectious bone disease, in addition to the parent drugs of these conjugates (ciprofloxacin (C), moxifloxacin (X) and tedizolid (T), Figure 2A) for direct comparison with our conjugates in preventing or eradicating planktonic and biofilm osteomyelitis pathogens in vitro and ex vivo.
2.2.1. Antimicrobials tested.
The following antibiotics (Figure 2A) were tested: minocycline (M), ciprofloxacin (C), moxifloxacin (X), tedizolid (T), and vancomycin (V); the following experimental conjugates were tested: BCC, BCX, BCT and BET. These conjugates were prepared as described in our published work (Ebetino, Sun, McKenna, Sadrerafi & Cherian, 2020; Sedghizadeh et al., 2017b; Sedghizadeh et al., 2020). All antimicrobials were tested initially in standard susceptibility studies with the protocol, as detailed in the reference (Sedghizadeh et al., 2017b). Based on the results and efficacy data from these initial studies, various compounds were chosen for further testing in biofilm preventative and eradication experiments using HA.
2.2.2. Antimicrobial susceptibility testing.
All tested microbial strains (S. aureus and Aa) were screened for their sensitivity to the antibiotics and conjugates listed above per standard microdilution methods described in EUCAST guidelines and detailed in reference (Sedghizadeh et al., 2017b). Minimum inhibitory concentrations (MIC90) were calculated and refer to a specific concentration of antimicrobial where 90% of bacterial growth was inhibited. For biofilm experiments, the MBEC90 (minimum biofilm eradication concentration), defined as the minimum concentration necessary to eradicate 90% of already formed biofilm, was calculated instead of the MIC90. The MIC90 and MBEC90 results for the conjugates (BCC, BCX, BCT, BET) were calculated based on the amount of their parent antibiotic to allow molar comparison with the parent drug. MIC90 and MBEC90 of tested antibiotics and conjugates against S. aureus and Aa strains is presented in Table 1.
Table 1.
Antimicrobial susceptibility results for experimental pathogens in polystyrene wells.
| Antimicrobials [μg/mL] | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M | C | X | T | V | BCC | BCX | BCT | BET | ||||||||||
| Microbial pathogens |
MIC | MBEC | MIC | MBEC | MIC | MBEC | MIC | MBEC | MIC | MBEC | MIC | MBEC | MIC | MBEC | MIC | MBEC | MIC | MBEC |
| S. aureus | 0.25 | 16 | 0.5 | 32 | 0.05 | 0.5 | 0.5 | 8 | 1 | 8 | 8 | 64 | 4 | 16 | 16 | 32 | 4 | 32 |
| Aa | 2 | 8 | 0.5 | 8 | 0.05 | 0.1 | * | * | * | * | 16 | 250 | 2 | 4 | * | * | * | * |
M – Minocycline; C – Ciprofloxacin; X – Moxifloxacin; T – Tedizolid; V – Vancomycin
spectrum of antimicrobial activity does not cover this specific pathogen and not supported for use against specific bacteria. For experimental purposes, the following reference strains were used: S. aureus ATCC 6538 and Aggregatibacter actinomycetemcomitans (Aa) D7S1. Moreover, 27 clinical strains of S. aureus were screened for their susceptibility to tested antibiotics (adapted from ref. (Ebetino, Sun, McKenna, Sadrerafi & Cherian, 2020; Sedghizadeh et al., 2020)).
2.2.3. Affinity of tested compounds to HA (Ebetino, Sun, McKenna, Sadrerafi & Cherian, 2020; Sedghizadeh et al., 2020).
Having established reference values for MIC90 and MBEC90, we evaluated the relative binding affinity of tested antibiotics and conjugates for HA powder. 1 μg/mL of each compound was added to an aqueous solution containing 10 μg/mL of HA powder (pH 6.7 - 6.9) and incubated for 4h/37°C under magnetic stirring. Next, HA powder was allowed to sediment for 1h/4°C, and the unbound antibiotic in the supernatant was measured using HPLC (Sedghizadeh et al., 2017b). 1 μg/mL of each compound incubated at the same condition, but without HA, served as a control sample. Affinity of compounds for HA powder was calculated as follows: 100% - peak area of tested compound detected/peak area of control sample *100%. The results obtained were expressed as [%] bound and were as follows: BET=92.7 ≈ BCT=92.1 ≈ BCX=91.8 ≈ BCC=91.2 > M=49.1 > X=40.3 ≈ C=36.2 > V=28.2 > T=23.6. The highest affinity for HA was seen with these BP-antibiotic conjugates as compared to the non-conjugated or parent antibiotics alone. Of the individual non-conjugated antibiotics, minocycline demonstrated the highest affinity for HA. Tedizolid demonstrated the lowest affinity for HA, but when conjugated with BP also demonstrated very high affinity.
2.2.4. Infection prevention experiment with HA.
To assess the ability of the conjugates and antibiotics to prevent bone infection in an in vitro model, we saturated HA spherules with concentrations of compounds equal to the MIC90 for each pathogen, removed the non-adherent compounds by three cycles of centrifugation-rinsing-decanting, and then introduced a solution containing pathogens to this experimental setting (Sedghizadeh et al., 2017b). Results are expressed as Planktonic Survival Rate [%] for the MIC90 values of compounds tested against S. aureus and Aa, as shown in Figure 3 (A-B). In this HA experimental setting against S. aureus, all of the BP conjugates (BCC, BET, BCX), but none of the non-conjugated/parent antibiotics, demonstrated statistically significant (K-W test, p<0.05) efficacy in preventing bacterial growth when compared to controls. Against Aa, only the BCX prevented bacterial growth at a statistically significant level (K-W test, p<0.05) compared to controls.
FIGURE 3.
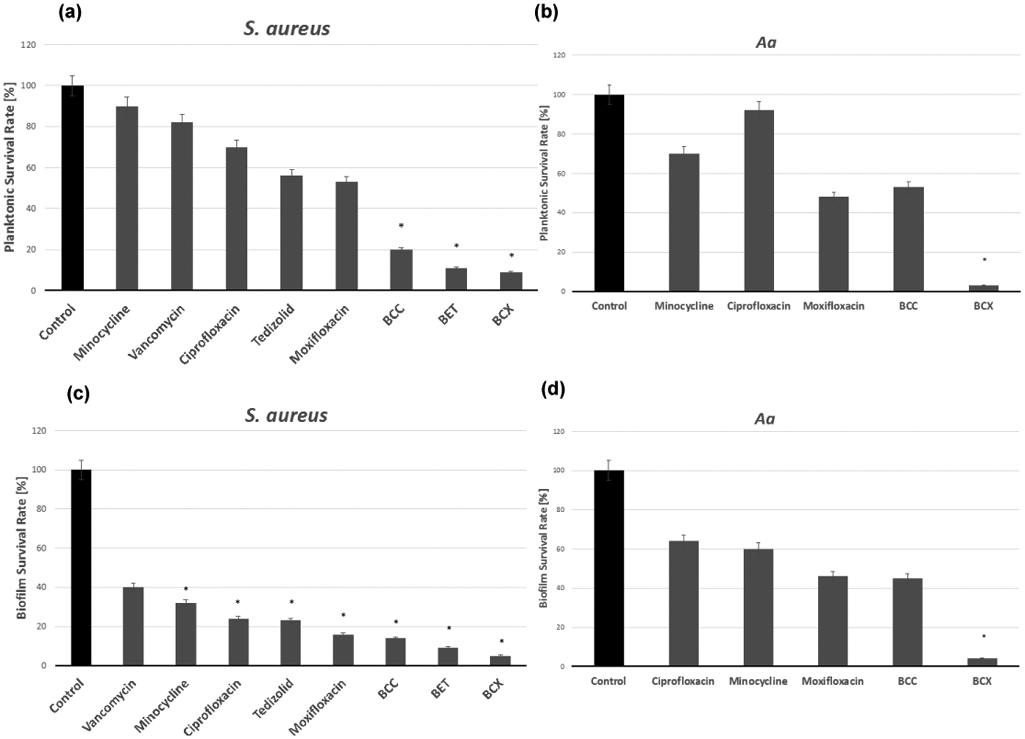
(a, b) Preventative hydroxyapatite experiments with tested compounds against S. aureus and Aa. (a) The BCC, BET, and BCX conjugates demonstrated statistically significant efficacy for preventing the growth of S. aureus. (b) The BCX conjugate demonstrated statistically significant efficacy for preventing the growth of Aa. (c, d) Eradication experiments with test compounds against S. aureus and Aa biofilms on HA. (c) All tested compounds except vancomycin were significantly efficacious in eradicating S. aureus, with the greatest reduction seen with BCX. (d) The BCX eradicated a significant number of Aa cells. * P<0.05, significantly different from control. Figure adapted from Ebetino et al., 2020; Sedghizadeh et al., 2021)
2.2.5. Biofilm eradication experiment with HA.
In this experimental setting, we looked for differences in the ability of the conjugates and non-BP-conjugated antibiotics to penetrate through established biofilms and the underlying matrix to inhibit microbial cells. 5ml containing 105 CFU/mL of each tested pathogen was introduced to HA pellets and the samples were subjected to incubation for 24h/37°C under magnetic stirring. Next, specific concentrations of tested compounds were added to samples (n=10 for each pathogen) in a manner that their final concentration was equivalent to the MBEC90 for biofilm forms of pathogens, as determined above. The subsequent procedures of incubation (5h/37°C), centrifugation, and spectrometric analysis utilizing a TTC assay were performed, as described elsewhere (Dydak et al., 2018). Results are expressed as Biofilm Survival Rate [%] for the MBEC90 values of compounds tested against S. aureus and Aa as shown in Figure 3 (C-D). In this HA experimental setting, several antibiotics and conjugates (BCC, BET, BCX) demonstrated statistically significant (K-W test, p<0.05) efficacy in eradicating established S. aureus biofilms compared to the controls, with the greatest relative efficacy seen with the conjugates compared to the non-conjugated antibiotics. Against Aa, only the BCX demonstrated statistically significant (K-W test, p<0.05) efficacy in eradicating biofilms when compared to controls.
2.2.6. Ex vivo bone biofilm experiment.
Prevention and eradication experiments utilizing bones (rat femurs and jaws) were performed in a manner analogous to those where HA was used, as described in the sections above for S. aureus and Aa, and again the survival rate of pathogens subjected to antimicrobials was calculated. The only differences were that no magnetic stirring was applied and experiments were carried out in 6-well plates. The key considerations of this experiment are: 1) The number of untreated cells (not exposed to antimicrobials) was considered 100%; survival rate was calculated as follows: the number of treated cells/number of untreated cells*100%; 2) Since the process of bone cleansing could introduce bacterial/fungal contamination, sterilization was required before adding the microbial inoculum to bone. To confirm efficacy of sterilization, testing of bone sterility was performed (as described in Figure 4 caption); 3) After cleansing and sterilization, bones were kept at −80°C in sterile containers to prevent their decomposition and microbial contamination. Bones were taken out of −80°C immediately before all procedures related to biofilm culturing; 4) Bones were used as a surface for biofilm growth (culture S. aureus on femoral bones and Aa on jawbones). Since use of bone samples with high variance in weight could lead to non-comparable results with regard to the number of biofilm-forming colonies, we decided to use only those bone samples that did not differ significantly and established the weight of bone as a basic criterion for similarity. The average weight of cleansed rat femora and jaws was 0.1901g and 0.3213g, respectively. For further experiments, only these bones for which the weight differed by less than 10% were used. However, it should be noted that due to differences in bone shapes and weights in individual rat anthropometrics, results obtained in this section only serve as a proof of principle of higher affinity to bone and antimicrobial activity of the conjugates in the bone environment in comparison to commonly used antibiotics. These findings are consistent with our in vitro results presented herein. S. aureus was chosen for experiments performed on long bones as this is the most common long bone osteomyelitis pathogen, and Aa was chosen for experiments performed on rat jaw since this is a common jawbone osteomyelitis pathogen. Results of preventative and eradication experiments on ex vivo bones are shown in Figure 4. In S. aureus long bone biofilm prevention and eradication settings ex vivo, the greatest efficacy was seen with the BET in both settings and tedizolid was also highly effective at eradicating established biofilms. In Aa jawbone biofilm prevention and eradication settings ex vivo, significant efficacy was only seen with the BCC in both settings (K-W test, p<0.05).
Figure 4.

Ex vivo assays of rat femora saturated with antimicrobials against S. aureus (A, B) and rat jaws saturated with antimicrobials against Aa (C, D). A. A picture of rat femora saturated with antibiotics or conjugates after they are introduced to a solution of S. aureus in our ex vivo preventative experimental setting. The more the bone appears red, the more staphylococcal biofilms there are adhered to the surface in those regions. Femur 1 – negative control sample (no antimicrobial, no staphylococcus); 2 – positive control sample (no antimicrobial, staphylococcal solution added); 3 – bone saturated with tedizolid and introduced to Staphylococcal solution; 4 – bone saturated with ciprofloxacin and introduced to staphylococcal solution; 5 – bone saturated with BET conjugate and introduced to staphylococcal solution; 6 – bone saturated with BCC and introduced to staphylococcal solution. B. In S. aureus biofilm prevention and eradication settings, conjugates BCC and BET significantly prevented or eradicated biofilm growth on femurs ex vivo, as did tedizolid alone, with greatest efficacy seen with the BET in both settings (K-W test, * p<0.05 versus control). C. A picture of rat jaws saturated with antibiotics or conjugates after they are introduced to a solution of Aa in our ex vivo preventative experimental setting. The more the bone appears red, the more Aa biofilms there are adhered to the surface in those regions. Jaw 1 – negative control sample (no antimicrobial, no Aa); 2 – positive control sample (no antimicrobial, Aa solution added); 3 – bone saturated with BCC and introduced to Aa solution; 4 – bone saturated with ciprofloxacin and introduced to Aa solution; 5 – bone saturated with minocycline and introduced to Aa solution. D. In Aa biofilm prevention and eradication settings, the BCC significantly prevented biofilm growth on jaws ex vivo whereas ciprofloxacin and minocycline were less effective in both settings (K-W test, * p<0.05; comparator=control). (Rats were killed by intraperitoneal application of pentobarbital (40 mg/kg), the chest was surgically opened and the heart was removed. No drugs or other procedures, which could potentially influence bone structure, were performed during rat inbreeding or experiments. The jaws were removed postmortem from Wistar male rats weighing 300-350g. To obtain osseous surfaces for biofilm development, the soft tissues were surgically removed from their bones after resection. Subsequently, bones were antiseptically rinsed using saline and then UV-irradiated to achieve sterilization. To test bone sterility, 6 bones (3 jaws and 3 femora) were incubated in anaerobic conditions in Thioglycolate Broth for 7 days according to microbiological procedures of anaerobic organism culturing. Another 6 bones (3 jaws and 3 femora) were incubated for 3 days in aerobic conditions in Brain-Heart Infusion (BHI) Broth. The sterilized bones were then frozen at −80°C to prevent their decomposition and microbial contamination). (Figure adapted from ref. (Ebetino, Sun, McKenna, Sadrerafi & Cherian, 2020; Sedghizadeh et al., 2019; Sedghizadeh, 2019; Sedghizadeh et al., 2021)).
2.2.7. Discussion.
In standard antimicrobial susceptibility testing (planktonic cultures and biofilm cultures grown on polystyrene), we found that each tested antibiotic or parent drug was more efficacious than the conjugates BCC, BCX, BCT and BET against long bone (S. aureus) and jawbone (Aa) osteomyelitis pathogens, as supported by MIC90 and MBEC90 data (Table 1). However, when HA was used instead of polystyrene to grow the same planktonic pathogens or biofilms, the tested conjugates were more efficacious in both infection prevention and treatment experiments (Figure 3). Similar results were also observed from the ex vivo experiments using rat femoral and jaw bones (Figure 4). This is most likely due to the stronger bone affinity of the conjugates, and thus higher local concentrations of antibiotic in the infected bone compartment compared to non-BP-conjugated antibiotics, supported by the HA affinity assays (section 2.2.3). Since BPs are known to target high turnover sites of the skeleton, these concentrations are further magnified under biofilms where bone resorption is known to occur. This bone binding and retention with sustained release of antibiotic over time confers advantages to the conjugates in this setting as compared to antibiotics without BP conjugation. Of the non-BP-conjugated antibiotics, minocycline demonstrated the greatest affinity to HA, which is consistent with the known relatively high bone affinity of the tetracycline class in general, as compared to other antibiotic classes. These findings lend support to the common clinical practice in dentistry of using minocycline, often topically, for treatment of jawbone infections, such as periodontitis, osteomyelitis, and osteonecrosis (Karasneh, Al-Eryani, Clark & Sedghizadeh, 2016).
The mechanism of action of individual antibiotics, such as those tested here, is well-known (Aldred, Kerns & Osheroff, 2014; Kisgen, Mansour, Unger & Childs, 2014). However, the exact mechanisms of activity for the novel conjugates remain to be elucidated. Conjugation is a chemical modification that can alter the biochemical interactions of the antibiotic prior to release from the BP moiety. As a result, properties of the parent drug, including its pharmacodynamics, can be altered by such modification until released. It has been shown in previous studies that conjugates in this class can retain the antibiotic activity of the parent drug (Herczegh et al., 2002), albeit at lower levels, which we also confirmed here. However, our non-cleavable BAC analog did not show any significant activity against osteomyelitis biofilms grown on HA surfaces in vitro (Sedghizadeh et al., 2017b) (Table 1). Also, as discussed above, a single dose of BCC was more efficacious than multiple doses of ciprofloxacin in vivo (Figure 2B). Together with the additional in vitro and ex vivo preventative/eradication experiments discussed above (Figure 3, 4), it is likely that release of antibiotic at the infected sites occurs, and these conjugates have a likely depot effect providing a longer term release of antibiotic at infected sites. Bacteria at those sites should therefore be subjected to a relatively higher and sustained concentration of the antibiotic. We are making progress at magnifying this effect, as we have demonstrated enhanced efficacy with conjugates, based on more potent fluoroquinolones. Indeed, the in vitro HA biofilm prevention and eradication results presented herein indicated that the most efficacious conjugate (BCX) against both S. aureus and Aa pathogens is also derived from the more potent fluoroquinolones tested (Table 1, Figure 3). In addition, the release rate is also a critical feature, as faster release could reduce the depot benefit, but slow release could reduce the critical local concentration at any given time point. We have observed in the MIC90 assays against S. aureus on polystyrene (Table 1) that an ester-linked BP-antibiotic conjugate (BET) leads to greater in vitro antimicrobial activity than a carbamate-linked conjugate (BCT), presumably due to faster release kinetics associated with ester hydrolysis as compared to carbamate linkages. Whether this will translate to greater potency in vivo remains to be studied.
These recent data show that this class of drug-releasing conjugates, incorporating osteo-adsorptive BPs with high bone affinity and fluoroquinolone or oxazolidinone antibiotics for bone-targeted delivery to treat osteomyelitis biofilm pathogens, constitutes an effective and promising approach to providing higher antimicrobial potency than unconjugated antimicrobial agents at high turnover bone sites, while minimizing systemic exposure and toxicity. In addition, bone affinity, antibiotic potency, and conjugation schemes are important for antimicrobial efficacy against osteomyelitis pathogens because they impact bone binding, antibiotic activity and release. These findings will aid the chemistry optimization for antimicrobial effects in future iterations of conjugates in this class and offer bisphosphonates and useful linkages for use in the design of new selective therapies for other bone-related diseases.
SUMMARY
An ideal therapeutic drug for bone diseases should act only on bone tissue with no pharmacological activity at other anatomical sites, as a further refinement of the “magic bullet” concept formulated by Ehrlich in 1900 (Tan & Grimes, 2010). Even more ideal is a drug that targets the most diseased sites of the skeleton, which is an attribute that BPs provide with their propensity to favor higher bone turnover sites. Research into bone-seeking medicinal agents, such as the work presented here is progressively laying the foundation for next-generation BP “target and release” type 'magic bullets' that minimize systemic exposure or toxicity and maximize drug efficacy at the targeted site.
ACKNOWLEDGEMENTS
This work was supported by grants (R41DE025789, R42DE025789, R43AI125060 and R43AR073727, R21AR069789, R21AR070984, R01AR063650) from the National Institutes of Health (NIDCR, NIAID, NIAMS).
ABBREVIATIONS
- (Aa)
Aggregatibacter actinomycetemcomitans
- (BP)
Bisphosphonate
- (BM)
Bone marrow
- (Btz)
Bortezomib
- (BR)
Bortezomib resistance
- (BHI)
Brain-Heart Infusion
- (CCK8)
Cell counting kit-8
- (CQ)
Chloroquine
- (C)
Ciprofloxacin
- (EUCAST)
European Committee on Antimicrobial Susceptibility Testing
- (HA)
Hydroxyapatite
- (HCQ)
Hydroxychloroquine
- (MSSA)
Methicillin-susceptible Staphylococcus aureus
- (MRSA)
Methicillin-resistant Staphylococcus aureus
- (MIC90)
Minimum inhibitory concentrations where 90% of bacterial growth was inhibited
- (MBEC90)
Minimum biofilm eradication concentration necessary to eradicate 90% of already formed biofilm
- (M)
Minocycline
- (X)
Moxifloxacin
- (MM)
Multiple myeloma
- (MLC1,2)
Myosin light chain 1,2
- (NPs)
Nano particles
- (PJI)
Prosthetic joint infection
- (Runx2)
Runt Related Transcription Factor 2
- (T)
Tedizolid
- (V)
Vancomycin
Footnotes
Nomenclature of Targets and Ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander et al., 2019).
DISCLOSURE
S.S., P.C., and F.H.E are employees of BioVinc LLC; S.S., F.H.E., and C.E.M. are founders and shareholders of BioVinc LLC; P.P. and J.D.N. are consultants to BioVinc LLC; RKB is a consultant for Novartis. The other authors do not have disclosures.
REFERENCES
- Abdel Malek MA, Jagannathan S, Malek E, Sayed DM, Elgammal SA, Abd El-Azeem HG, et al. (2015). Molecular chaperone GRP78 enhances aggresome delivery to autophagosomes to promote drug resistance in multiple myeloma. Oncotarget 6: 3098–3110. doi: 10.18632/oncotarget.3075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrahamsen B (2010). Adverse effects of bisphosphonates. Calcif Tissue Int 86: 421–435. doi: 10.1007/s00223-010-9364-1 [DOI] [PubMed] [Google Scholar]
- Agyin JK, Santhamma B, & Roy SS (2013). Design, synthesis, and biological evaluation of bone-targeted proteasome inhibitors for multiple myeloma. Bioorg Med Chem Lett 23: 6455–6458. doi: 10.1016/j.bmcl.2013.09.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Farsi K (2013). Multiple Myeloma: An Update. Oman Medical Journal 28. doi: 10.5001/omj.2013.02 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldred KJ, Kerns RJ, & Osheroff N (2014). Mechanism of quinolone action and resistance. Biochemistry 53: 1565–1574. doi: 10.1021/bi5000564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Mathie A, Peters JA, Veale EL, et al. (2019). THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Enzymes. Br J Pharmacol 176 Suppl 1: S297–S396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attar-Schneider O, Drucker L, Zismanov V, Tartakover-Matalon S, Rashid G, & Lishner M (2012). Bevacizumab attenuates major signaling cascades and eIF4E translation initiation factor in multiple myeloma cells. Lab Invest 92: 178–190. doi: 10.1038/labinvest.2011.162 [DOI] [PubMed] [Google Scholar]
- Auner HW, & Cenci S (2015). Recent advances and future directions in targeting the secretory apparatus in multiple myeloma. Br J Haematol 168: 14–25. doi: 10.1111/bjh.13172 [DOI] [PubMed] [Google Scholar]
- Boeckman RK Jr., Boyce BF, Xiao L, YAO Z, & Ebetino FH (2017). Phosphonate-chloroquine conjugates and methods using same. [Google Scholar]
- Boelch SP, Rueckl K, Fuchs C, Jordan M, Knauer M, Steinert A, et al. (2018). Comparison of Elution Characteristics and Compressive Strength of Biantibiotic-Loaded PMMA Bone Cement for Spacers: Copal(R) Spacem with Gentamicin and Vancomycin versus Palacos(R) R+G with Vancomycin. Biomed Res Int 2018: 4323518. doi: 10.1155/2018/4323518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Both T, Zillikens MC, Schreuders-Koedam M, Vis M, Lam WK, Weel A, et al. (2018). Hydroxychloroquine affects bone resorption both in vitro and in vivo. J Cell Physiol 233: 1424–1433. doi: 10.1002/jcp.26028 [DOI] [PubMed] [Google Scholar]
- Boyce BF (2019). Delivery of chloroquine and anti-cancer agents to bone. JBMR plus 3: 14.30680359 [Google Scholar]
- Bozhkova SA, Novokshonova AA, & Konev VA (2015). Current trends in local antibacterial therapy of periprosthetic infection and osteomyelitis. Travmatol Ortop Ross: 92–107. doi: 10.21823/2311-2905-2015-0-3-92-107 [DOI] [Google Scholar]
- Bozic KJ, Kamath AF, Ong K, Lau E, Kurtz S, Chan V, et al. (2015). Comparative Epidemiology of Revision Arthroplasty: Failed THA Poses Greater Clinical and Economic Burdens Than Failed TKA. Clin Orthop Relat Res 473: 2131–2138. doi: 10.1007/s11999-014-4078-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer JW, & Diehl JA (2000). PERK mediates cell-cycle exit during the mammalian unfolded protein response. Proc Natl Acad Sci U S A 97: 12625–12630. doi: 10.1073/pnas.220247197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning DJ (2014). Pharmacology of Chloroquine and Hydroxychloroquine. In Hydroxychloroquine and Chloroquine Retinopathy. ed Browning DJ Springer New York: New York, NY, pp 35–63, DOI: 10.1007/978-1-4939-0597-3_2. [DOI] [Google Scholar]
- Carreno JJ, Kenney RM, & Lomaestro B (2014). Vancomycin-associated renal dysfunction: where are we now? Pharmacotherapy 34: 1259–1268. doi: 10.1002/phar.1488 [DOI] [PubMed] [Google Scholar]
- Cavaletti G, Gilardini A, Canta A, Rigamonti L, Rodriguez-Menendez V, Ceresa C, et al. (2007). Bortezomib-induced peripheral neurotoxicity: a neurophysiological and pathological study in the rat. Exp Neurol 204: 317–325. doi: 10.1016/j.expneurol.2006.11.010 [DOI] [PubMed] [Google Scholar]
- Chauhan D, Singh AV, Aujay M, Kirk CJ, Bandi M, Ciccarelli B, et al. (2010). A novel orally active proteasome inhibitor ONX 0912 triggers in vitro and in vivo cytotoxicity in multiple myeloma. Blood 116: 4906–4915. doi: 10.1182/blood-2010-04-276626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen AT, & Vallier HA (2016). Noncontiguous and open fractures of the lower extremity: Epidemiology, complications, and unplanned procedures. Injury 47: 742–747. doi: 10.1016/j.injury.2015.12.013 [DOI] [PubMed] [Google Scholar]
- Cheong S, Sun S, Kang B, Bezouglaia O, Elashoff D, McKenna CE, et al. (2014). Bisphosphonate uptake in areas of tooth extraction or periapical disease. J Oral Maxillofac Surg 72: 2461–2468. doi: 10.1016/j.joms.2014.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HR, & Bedair H (2014). Mortality following revision total knee arthroplasty: a matched cohort study of septic versus aseptic revisions. J Arthroplasty 29: 1216–1218. doi: 10.1016/j.arth.2013.11.026 [DOI] [PubMed] [Google Scholar]
- Choi HR, Beecher B, & Bedair H (2013). Mortality after septic versus aseptic revision total hip arthroplasty: a matched-cohort study. J Arthroplasty 28: 56–58. doi: 10.1016/j.arth.2013.02.041 [DOI] [PubMed] [Google Scholar]
- Bortezomib clinicaltrials. [Online] Available from https://clinicaltrials.gov/ct2/results/browse?term=bortezomib&brwse=cond_alpha_all. [Accessed]. [Google Scholar]
- Cole LE, Vargo-Gogola T, & Roeder RK (2016). Targeted delivery to bone and mineral deposits using bisphosphonate ligands. Adv Drug Deliv Rev 99: 12–27. doi: 10.1016/j.addr.2015.10.005 [DOI] [PubMed] [Google Scholar]
- Corona PS, Espinal L, Rodriguez-Pardo D, Pigrau C, Larrosa N, & Flores X (2014). Antibiotic susceptibility in gram-positive chronic joint arthroplasty infections: increased aminoglycoside resistance rate in patients with prior aminoglycoside-impregnated cement spacer use. J Arthroplasty 29: 1617–1621. doi: 10.1016/j.arth.2014.03.029 [DOI] [PubMed] [Google Scholar]
- Costedoat-Chalumeau N, Hulot JS, Amoura Z, Delcourt A, Maisonobe T, Dorent R, et al. (2007). Cardiomyopathy related to antimalarial therapy with illustrative case report. Cardiology 107: 73–80. doi: 10.1159/000094079 [DOI] [PubMed] [Google Scholar]
- de Mesy Bentley KL, Trombetta R, Nishitani K, Bello-Irizarry SN, Ninomiya M, Zhang L, et al. (2017). Evidence of Staphylococcus Aureus Deformation, Proliferation, and Migration in Canaliculi of Live Cortical Bone in Murine Models of Osteomyelitis. J Bone Miner Res 32: 985–990. doi: 10.1002/jbmr.3055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dispenzieri A, Jacobus S, Vesole DH, Callandar N, Fonseca R, & Greipp PR (2010). Primary therapy with single agent bortezomib as induction, maintenance and re-induction in patients with high-risk myeloma: results of the ECOG E2A02 trial. Leukemia 24: 1406–1411. doi: 10.1038/leu.2010.129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubost JJ, Couderc M, Tatar Z, Tournadre A, Lopez J, Mathieu S, et al. (2014). Three-decade trends in the distribution of organisms causing septic arthritis in native joints: single-center study of 374 cases. Joint Bone Spine 81: 438–440. doi: 10.1016/j.jbspin.2014.05.001 [DOI] [PubMed] [Google Scholar]
- Dydak K, Junka A, Szymczyk P, Chodaczek G, Toporkiewicz M, Fijalkowski K, et al. (2018). Development and biological evaluation of Ti6Al7Nb scaffold implants coated with gentamycin-saturated bacterial cellulose biomaterial. PLoS One 13: e0205205. doi: 10.1371/journal.pone.0205205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dykstra KM, Allen C, Born EJ, Tong H, & Holstein SA (2015). Mechanisms for autophagy modulation by isoprenoid biosynthetic pathway inhibitors in multiple myeloma cells. Oncotarget 6: 41535–41549. doi: 10.18632/oncotarget.6365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebetino FH, Hogan AM, Sun S, Tsoumpra MK, Duan X, Triffitt JT, et al. (2011). The relationship between the chemistry and biological activity of the bisphosphonates. Bone 49: 20–33. doi: 10.1016/j.bone.2011.03.774 [DOI] [PubMed] [Google Scholar]
- Ebetino FH, Sun S, McKenna CE, Sadrerafi K, & Cherian P (2020). Bone targeted antimicrobial oxazolidinone related compounds, formulations thereof, and uses thereof BioVinc LLC, p 112pp. WO2020005964A1, https://patents.google.com/patent/WO2020005964A1/en?oq=WO2020005964A1 [Google Scholar]
- El Arfani C, De Veirman K, Maes K, De Bruyne E, & Menu E (2018). Metabolic Features of Multiple Myeloma. Int J Mol Sci 19. doi: 10.3390/ijms19041200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell KB, Karpeisky A, Thamm DH, & Zinnen S (2018). Bisphosphonate conjugation for bone specific drug targeting. Bone Rep 9: 47–60. doi: 10.1016/j.bonr.2018.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrand J, El Samad Y, Brunschweiler B, Grados F, Dehamchia-Rehailia N, Sejourne A, et al. (2016). Morbimortality in adult patients with septic arthritis: a three-year hospital-based study. BMC Infect Dis 16: 239. doi: 10.1186/s12879-016-1540-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogelman I (1982). Diphosphonate bone scanning agents--current concepts. Eur J Nucl Med 7: 506–509. doi: 10.1007/bf00257217 [DOI] [PubMed] [Google Scholar]
- Fong IW, Ledbetter WH, Vandenbroucke AC, Simbul M, & Rahm V (1986). Ciprofloxacin concentrations in bone and muscle after oral dosing. Antimicrob Agents Chemother 29: 405–408. doi: 10.1128/AAC.29.3.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraimow HS (2009). Systemic antimicrobial therapy in osteomyelitis. Seminars in plastic surgery 23: 90–99. doi: 10.1055/s-0029-1214161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghobrial IM, Detappe A, Anderson KC, & Steensma DP (2018). The bone-marrow niche in MDS and MGUS: implications for AML and MM. Nat Rev Clin Oncol 15: 219–233. doi: 10.1038/nrclinonc.2017.197 [DOI] [PubMed] [Google Scholar]
- Giuliani N, Morandi F, Tagliaferri S, Lazzaretti M, Bonomini S, Crugnola M, et al. (2007). The proteasome inhibitor bortezomib affects osteoblast differentiation in vitro and in vivo in multiple myeloma patients. Blood 110: 334–338. doi: 10.1182/blood-2006-11-059188 [DOI] [PubMed] [Google Scholar]
- Giuliani N, & Rizzoli V (2007). Myeloma cells and bone marrow osteoblast interactions: Role in the development of osteolytic lesions in multiple myeloma. Leukemia & Lymphoma, 48, 2323–2329. doi: 10.1080/10428190701648281 [DOI] [PubMed] [Google Scholar]
- Goldschmidt H, Ashcroft J, Szabo Z, & Garderet L (2019). Navigating the treatment landscape in multiple myeloma: which combinations to use and when? Ann Hematol 98: 1–18. doi: 10.1007/s00277-018-3546-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal V, & Bordia A (1995). The hypoglycemic effect of chloroquine. J Assoc Physicians India 43: 17–18. [PubMed] [Google Scholar]
- Hamouda MA, Belhacene N, Puissant A, Colosetti P, Robert G, Jacquel A, et al. (2014). The small heat shock protein B8 (HSPB8) confers resistance to bortezomib by promoting autophagic removal of misfolded proteins in multiple myeloma cells. Oncotarget 5: 6252–6266. doi: 10.18632/oncotarget.2193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S, Gray AJG, Bruce L, Alexander SPH, Anderton S, Bryant C, Davenport AP, Doerig C, Fabbro D, Levi-Schaffer F, Spedding M, Davies JA, NC-IUPHAR (2018). The IUPHAR/BPS guide to pharmacology in 2018: Updates and expansion to encompass the new guide to immunopharmacology. Nucleic Acids Research 46, D1091–D1106. doi: 10.1093/nar/gkx1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herczegh P, Buxton TB, McPherson JC 3rd, Kovacs-Kulyassa A, Brewer PD, Sztaricskai F, et al. (2002). Osteoadsorptive bisphosphonate derivatives of fluoroquinolone antibacterials. J Med Chem 45: 2338–2341. doi: 10.1021/jm0105326 [DOI] [PubMed] [Google Scholar]
- Hideshima T, Ikeda H, Chauhan D, Okawa Y, Raje N, Podar K, et al. (2009). Bortezomib induces canonical nuclear factor-kappaB activation in multiple myeloma cells. Blood 114: 1046–1052. doi: 10.1182/blood-2009-01-199604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang B, Benavides A, Shi Y, Frost P, & Lichtenstein A (2009). Effect of autophagy on multiple myeloma cell viability. Mol Cancer Ther 8: 1974–1984. doi: 10.1158/1535-7163.MCT-08-1177 [DOI] [PubMed] [Google Scholar]
- Jin J, Okamoto R, Yoon SS, Shih LY, Zhu J, Liu T, et al. (2018). Bortezomib-based therapy for transplant-ineligible East Asian patients with newly diagnosed mantle-cell lymphoma. Onco Targets Ther 11: 3869–3882. doi: 10.2147/OTT.S150339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junka A, Szymczyk P, Ziolkowski G, Karuga-Kuzniewska E, Smutnicka D, Bil-Lula I, et al. (2017). Bad to the Bone: On In Vitro and Ex Vivo Microbial Biofilm Ability to Directly Destroy Colonized Bone Surfaces without Participation of Host Immunity or Osteoclastogenesis. PLoS One 12: e0169565. doi: 10.1371/journal.pone.0169565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath AF, Ong KL, Lau E, Chan V, Vail TP, Rubash HE, et al. (2015). Quantifying the Burden of Revision Total Joint Arthroplasty for Periprosthetic Infection. J Arthroplasty 30: 1492–1497. doi: 10.1016/j.arth.2015.03.035 [DOI] [PubMed] [Google Scholar]
- Karasneh JA, Al-Eryani K, Clark GT, & Sedghizadeh PP (2016). Modified protocol including topical minocycline in orabase to manage medication-related osteonecrosis of the jaw cases. J Oral Pathol Med 45: 718–720. doi: 10.1111/jop.12419 [DOI] [PubMed] [Google Scholar]
- Kawano Y, Moschetta M, Manier S, Glavey S, Gorgun GT, Roccaro AM, et al. (2015). Targeting the bone marrow microenvironment in multiple myeloma. Immunol Rev 263: 160–172. doi: 10.1111/imr.12233 [DOI] [PubMed] [Google Scholar]
- Keller DL (2014). Adverse cardiac effects of bisphosphonates. Mayo Clin Proc 89: 1025–1026. doi: 10.1016/j.mayocp.2014.04.012 [DOI] [PubMed] [Google Scholar]
- Kempfle JS, Nguyen K, Hamadani C, Koen N, Edge AS, Kashemirov BA, et al. (2018). Bisphosphonate-Linked TrkB Agonist: Cochlea-Targeted Delivery of a Neurotrophic Agent as a Strategy for the Treatment of Hearing Loss. Bioconjug Chem 29: 1240–1250. doi: 10.1021/acs.bioconjchem.8b00022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BN, Kim ES, & Oh MD (2014). Oral antibiotic treatment of staphylococcal bone and joint infections in adults. J Antimicrob Chemother 69: 309–322. doi: 10.1093/jac/dkt374 [DOI] [PubMed] [Google Scholar]
- Kisgen JJ, Mansour H, Unger NR, & Childs LM (2014). Tedizolid: a new oxazolidinone antimicrobial. Am J Health Syst Pharm 71: 621–633. doi: 10.2146/ajhp130482 [DOI] [PubMed] [Google Scholar]
- Kowada T, Kikuta J, Kubo A, Ishii M, Maeda H, Mizukami S, et al. (2011). In vivo fluorescence imaging of bone-resorbing osteoclasts. J Am Chem Soc 133: 17772–17776. doi: 10.1021/ja2064582 [DOI] [PubMed] [Google Scholar]
- Kuhn DJ, Berkova Z, Jones RJ, Woessner R, Bjorklund CC, Ma W, et al. (2012). Targeting the insulin-like growth factor-1 receptor to overcome bortezomib resistance in preclinical models of multiple myeloma. Blood 120: 3260–3270. doi: 10.1182/blood-2011-10-386789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavrador P, Gaspar VM, & Mano JF (2018). Stimuli-responsive nanocarriers for delivery of bone therapeutics - Barriers and progresses. J Control Release 273: 51–67. doi: 10.1016/j.jconrel.2018.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson MA, McDonald MM, Kovacic N, Hua Khoo W, Terry RL, Down J, et al. (2015). Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nat Commun 6: 8983. doi: 10.1038/ncomms9983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew DP, & Waldvogel FA (2004). Osteomyelitis. Lancet 364: 369–379. doi: 10.1016/S0140-6736(04)16727-5 [DOI] [PubMed] [Google Scholar]
- Li C, Chen S, Yue P, Deng X, Lonial S, Khuri FR, et al. (2010). Proteasome inhibitor PS-341 (bortezomib) induces calpain-dependent IkappaB(alpha) degradation. J Biol Chem 285: 16096–16104. doi: 10.1074/jbc.M109.072694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin B, Caldwell C, Bhaduri S, Goel V, & Agarwal A (2017). Optimizing Vancomycin Release from Calcium Phosphate-Based Cement by Carboxymethyl Cellulose for Prevention of Osteomyelitis. Surg Infect (Larchmt) 18: 221–222. doi: 10.1089/sur.2016.189 [DOI] [PubMed] [Google Scholar]
- Lonial S, Waller EK, Richardson PG, Jagannath S, Orlowski RZ, Giver CR, et al. (2005). Risk factors and kinetics of thrombocytopenia associated with bortezomib for relapsed, refractory multiple myeloma. Blood 106: 3777–3784. doi: 10.1182/blood-2005-03-1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahindra A, Hideshima T, & Anderson KC (2010). Multiple myeloma: biology of the disease. Blood Rev 24 Suppl 1: S5–11. doi: 10.1016/S0268-960X(10)70003-5 [DOI] [PubMed] [Google Scholar]
- Manasanch EE, & Orlowski RZ (2017). Proteasome inhibitors in cancer therapy. Nat Rev Clin Oncol 14: 417–433. doi: 10.1038/nrclinonc.2016.206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna CE, Haratipour P, Duro MVV, & Ebetino FH (2020). Chemistry of Bisphosphonates. In Encyclopedia of Bone Biology. ed Zaidi M. Academic Press: Oxford, pp 551–564. DOI: 10.1016/B978-0-12-801238-3.11260-7. [DOI] [Google Scholar]
- Mitra AK, Mukherjee UK, Harding T, Jang JS, Stessman H, Li Y, et al. (2016). Single-cell analysis of targeted transcriptome predicts drug sensitivity of single cells within human myeloma tumors. Leukemia 30: 1094–1102. doi: 10.1038/leu.2015.361 [DOI] [PubMed] [Google Scholar]
- Montanari F, Lu M, Marcus S, Saran A, Malankar A, & Mazumder A (2014). A Phase II Trial of Chloroquine in Combination with Bortezomib and Cyclophosphamide in Patients with Relapsed and Refractory Multiple Myeloma. Blood 124: 5775–5775. doi: 10.1182/blood.V124.21.5775.5775 [DOI] [Google Scholar]
- Murai K, Kowata S, Shimoyama T, Yashima-Abo A, Fujishima Y, Ito S, et al. (2014). Bortezomib induces thrombocytopenia by the inhibition of proplatelet formation of megakaryocytes. Eur J Haematol 93: 290–296. doi: 10.1111/ejh.12342 [DOI] [PubMed] [Google Scholar]
- Murray MY, Auger MJ, & Bowles KM (2014). Overcoming bortezomib resistance in multiple myeloma. Biochem Soc Trans 42: 804–808. doi: 10.1042/BST20140126 [DOI] [PubMed] [Google Scholar]
- Nagaratnam N, Chetiyawardana AD, & Rajiyah S (1978). Aplasia and leukaemia following chloroquine therapy. Postgrad Med J 54: 108–112. doi: 10.1136/pgmj.54.628.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niewerth D, Jansen G, Assaraf YG, Zweegman S, Kaspers GJ, & Cloos J (2015). Molecular basis of resistance to proteasome inhibitors in hematological malignancies. Drug Resist Updat 18: 18–35. doi: 10.1016/j.drup.2014.12.001 [DOI] [PubMed] [Google Scholar]
- Obeng EA, Carlson LM, Gutman DM, Harrington WJ Jr., Lee KP, & Boise LH (2006). Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 107: 4907–4916. doi: 10.1182/blood-2005-08-3531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painuly U, & Kumar S (2013). Efficacy of bortezomib as first-line treatment for patients with multiple myeloma. Clin Med Insights Oncol 7: 53–73. doi: 10.4137/CMO.S7764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paton-Hough J, Chantry AD, & Lawson MA (2015). A review of current murine models of multiple myeloma used to assess the efficacy of therapeutic agents on tumour growth and bone disease. Bone 77: 57–68. doi: 10.1016/j.bone.2015.04.004 [DOI] [PubMed] [Google Scholar]
- Pazianas M, Cooper C, Ebetino FH, & Russell RG (2010). Long-term treatment with bisphosphonates and their safety in postmenopausal osteoporosis. Ther Clin Risk Manag 6: 325–343. doi: 10.2147/tcrm.s8054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennisi A, Li X, Ling W, Khan S, Zangari M, & Yaccoby S (2009). The proteasome inhibitor, bortezomib suppresses primary myeloma and stimulates bone formation in myelomatous and nonmyelomatous bones in vivo. Am J Hematol 84: 6–14. doi: 10.1002/ajh.21310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollak AN, Jones AL, Castillo RC, Bosse MJ, MacKenzie EJ, & Group LS (2010). The relationship between time to surgical debridement and incidence of infection after open high-energy lower extremity trauma. J Bone Joint Surg Am 92: 7–15. doi: 10.2106/JBJS.H.00984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabha A (1996). Hypoglycemic effect of chloroquine. J Assoc Physicians India 44: 149–150. [PubMed] [Google Scholar]
- Reinholz MM, Zinnen SP, Dueck AC, Dingli D, Reinholz GG, Jonart LA, et al. (2010). A promising approach for treatment of tumor-induced bone diseases: utilizing bisphosphonate derivatives of nucleoside antimetabolites. Bone 47: 12–22. doi: 10.1016/j.bone.2010.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson PG, Barlogie B, Berenson J, Singhal S, Jagannath S, Irwin D, et al. (2003). A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med 348: 2609–2617. doi: 10.1056/NEJMoa030288 [DOI] [PubMed] [Google Scholar]
- Richardson PG, Sonneveld P, Schuster MW, Irwin D, Stadtmauer EA, Facon T, et al. (2005). Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med 352: 2487–2498. doi: 10.1056/NEJMoa043445 [DOI] [PubMed] [Google Scholar]
- Richardson PG, Xie W, Jagannath S, Jakubowiak A, Lonial S, Raje NS, et al. (2014). A phase 2 trial of lenalidomide, bortezomib, and dexamethasone in patients with relapsed and relapsed/refractory myeloma. Blood 123: 1461–1469. doi: 10.1182/blood-2013-07-517276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robak T, Huang H, Jin J, Zhu J, Liu T, Samoilova O, et al. (2015). Bortezomib-based therapy for newly diagnosed mantle-cell lymphoma. N Engl J Med 372: 944–953. doi: 10.1056/NEJMoa1412096 [DOI] [PubMed] [Google Scholar]
- Roy P, Sarkar UA, & Basak S (2018). The NF-kappaB Activating Pathways in Multiple Myeloma. Biomedicines 6. doi: 10.3390/biomedicines6020059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruschak AM, Slassi M, Kay LE, & Schimmer AD (2011). Novel proteasome inhibitors to overcome bortezomib resistance. J Natl Cancer Inst 103: 1007–1017. doi: 10.1093/jnci/djr160 [DOI] [PubMed] [Google Scholar]
- Russell RG, Watts NB, Ebetino FH, & Rogers MJ (2008). Mechanisms of action of bisphosphonates: similarities and differences and their potential influence on clinical efficacy. Osteoporos Int 19: 733–759. doi: 10.1007/s00198-007-0540-8 [DOI] [PubMed] [Google Scholar]
- Scott EC, Maziarz RT, Spurgeon SE, Medvedova E, Gajewski J, Reasor-Heard S, et al. (2017). Double autophagy stimulation using chemotherapy and mTOR inhibition combined with hydroxychloroquine for autophagy modulation in patients with relapsed or refractory multiple myeloma. Haematologica 102: e261–e265. doi: 10.3324/haematol.2016.162321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedghizadeh P, Ebetino FH, Sun S, Sadrerafi K, Junka A, Mahabady S, et al. (2017a). Bone targeted bisphosphonate-antibiotic conjugates for the treatment of osteomyelitis biofilms. Journal of Bone and Mineral Res 32: S285–S285. [Google Scholar]
- Sedghizadeh P, Sodagar E, Garabedian R, Wadia J, Ebetino FH, Cherian P, et al. (2019). Design and in vivo testing of novel bisphosphonate-fluoroquinolone conjugates chemisorbed to bone graft material. Journal of Bone and Mineral Res 34: S367–S368. [Google Scholar]
- Sedghizadeh PP (2019). Biofilm mediated infections in dentistry and the role of bisphosphonates in oral diseases and therapeutics JBMR Plus 3: 48. [Google Scholar]
- Sedghizadeh PP, Sun S, Junka AF, Richard E, Sadrerafi K, Mahabady S, et al. (2017b). Design, Synthesis, and Antimicrobial Evaluation of a Novel Bone-Targeting Bisphosphonate-Ciprofloxacin Conjugate for the Treatment of Osteomyelitis Biofilms. J Med Chem 60: 2326–2343. doi: 10.1021/acs.jmedchem.6b01615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedghizadeh PP, Sun S, Jones AC, Sodagar E, Cherian P, Chen C, et al. (2021). Bisphosphonates in Dentistry: Historical Perspectives, Adverse Effects, and Novel Applications. Bone, In press, doi.org: 10.1016/j.bone.2021.115933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shain KH, Dalton WS, & Tao J (2015). The tumor microenvironment shapes hallmarks of mature B-cell malignancies. Oncogene 34: 4673–4682. doi: 10.1038/onc.2014.403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi DS, Smith MC, Campbell RA, Zimmerman PW, Franks ZB, Kraemer BF, et al. (2014). Proteasome function is required for platelet production. J Clin Invest 124: 3757–3766. doi: 10.1172/JCI75247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver IA, Murrills RJ, & Etherington DJ (1988). Microelectrode studies on the acid microenvironment beneath adherent macrophages and osteoclasts. Exp Cell Res 175: 266–276. doi: 10.1016/0014-4827(88)90191-7 [DOI] [PubMed] [Google Scholar]
- Slane J, Gietman B, & Squire M (2018). Antibiotic elution from acrylic bone cement loaded with high doses of tobramycin and vancomycin. J Orthop Res 36: 1078–1085. doi: 10.1002/jor.23722 [DOI] [PubMed] [Google Scholar]
- Staats K, Sevelda F, Kaider A, Bohler C, Sigmund IK, Puchner SE, et al. (2017). The influence of antibiotic-loaded cement spacers on the risk of reinfection after septic two-stage hip revision surgery. Infection 45: 885–891. doi: 10.1007/s15010-017-1081-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoodley P, Ehrlich GD, Sedghizadeh PP, Hall-Stoodley L, Baratz ME, Altman DT, et al. (2011). Orthopaedic biofilm infections. Curr Orthop Pract 22: 558–563. doi: 10.1097/BCO.0b013e318230efcf [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui X, Chen R, Wang Z, Huang Z, Kong N, Zhang M, et al. (2013). Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis 4: e838. doi: 10.1038/cddis.2013.350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swami A, Reagan MR, Basto P, Mishima Y, Kamaly N, Glavey S, et al. (2014). Engineered nanomedicine for myeloma and bone microenvironment targeting. Proc Natl Acad Sci U S A 111: 10287–10292. doi: 10.1073/pnas.1401337111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan SY, & Grimes S (2010). Paul Ehrlich (1854-1915): man with the magic bullet. Singapore Med J 51: 842–843. [PubMed] [Google Scholar]
- Tao J, Srinivasan V, Zhou X, Ebetino FH, Boeckman RK Jr. , Boyce BF, et al. (2018). Bone-targeting Bortezomib significantly increases it efficacy in the treatment of human multiple myeloma in vitro and in vivo in mice. Journal of Bone and Mineral Res 33: 45. [Google Scholar]
- Terpos E, Ntanasis-Stathopoulos I, Gavriatopoulou M, & Dimopoulos MA (2018). Pathogenesis of bone disease in multiple myeloma: from bench to bedside. Blood Cancer J 8: 7. doi: 10.1038/s41408-017-0037-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimoto Y, & Shimizu S (2005). Another way to die: autophagic programmed cell death. Cell Death Differ 12 Suppl 2: 1528–1534. doi: 10.1038/sj.cdd.4401777 [DOI] [PubMed] [Google Scholar]
- Uskokovic V (2015). Nanostructured platforms for the sustained and local delivery of antibiotics in the treatment of osteomyelitis. Crit Rev Ther Drug Carr Syst 32: 1–59. doi: 10.1615/CritRevTherDrugCarrierSyst.2014010920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uy GL, Trivedi R, Peles S, Fisher NM, Zhang QJ, Tomasson MH, et al. (2007). Bortezomib inhibits osteoclast activity in patients with multiple myeloma. Clin Lymphoma Myeloma 7: 587–589. doi: 10.3816/CLM.2007.n.045 [DOI] [PubMed] [Google Scholar]
- Verbaanderd C, Maes H, Schaaf MB, Sukhatme VP, Pantziarka P, Sukhatme V, et al. (2017). Repurposing Drugs in Oncology (ReDO)-chloroquine and hydroxychloroquine as anti-cancer agents. Ecancermedicalscience 11: 781. doi: 10.3332/ecancer.2017.781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogl DT, Stadtmauer EA, Tan KS, Heitjan DF, Davis LE, Pontiggia L, et al. (2014). Combined autophagy and proteasome inhibition: a phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy 10: 1380–1390. doi: 10.4161/auto.29264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Metzler I, Krebbel H, Hecht M, Manz RA, Fleissner C, Mieth M, et al. (2007). Bortezomib inhibits human osteoclastogenesis. Leukemia 21: 2025–2034. doi: 10.1038/sj.leu.2404806 [DOI] [PubMed] [Google Scholar]
- Vrabel D, Pour L, & Sevcikova S (2019). The impact of NF-kappaB signaling on pathogenesis and current treatment strategies in multiple myeloma. Blood Rev 34: 56–66. doi: 10.1016/j.blre.2018.11.003 [DOI] [PubMed] [Google Scholar]
- Walker LC, Baker P, Holleyman R, & Deehan D (2017). Microbial resistance related to antibiotic-loaded bone cement: a historical review. Knee Surg Sports Traumatol Arthrosc 25: 3808–3817. doi: 10.1007/s00167-016-4309-5 [DOI] [PubMed] [Google Scholar]
- Wang H, Xiao L, Tao J, Srinivasan V, Boyce BF, Ebetino FH, et al. (2018). Synthesis of a Bone-Targeted Bortezomib with In Vivo Anti-Myeloma Effects in Mice. Pharmaceutics 10. doi: 10.3390/pharmaceutics10030154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Zhang H, Srinivasan V, Tao J, Sun W, Lin X, et al. (2020). Targeting Bortezomib to Bone Increases Its Bone Anabolic Activity and Reduces Systemic Adverse Effects in Mice. J Bone Miner Res 35: 343–356. doi: 10.1002/jbmr.3889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Cai X, Yang J, Wang C, Tong L, Xiao J, et al. (2018). A Targeted and pH-Responsive Bortezomib Nanomedicine in the Treatment of Metastatic Bone Tumors. ACS Appl Mater Interfaces 10: 41003–41011. doi: 10.1021/acsami.8b07527 [DOI] [PubMed] [Google Scholar]
- Xing L, Ebetino FH, Boeckman RK Jr., Srinivasan V, Tao J, Sawyer TK, et al. (2020). Targeting anti-cancer agents to bone using bisphosphonates. Bone 138: 115492. doi: 10.1016/j.bone.2020.115492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiu Y, Xu H, Zhao C, Li J, Morita Y, Yao Z, et al. (2014). Chloroquine reduces osteoclastogenesis in murine osteoporosis by preventing TRAF3 degradation. J Clin Invest 124: 297–310. doi: 10.1172/JCI66947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, De Veirman K, De Becker A, Vanderkerken K, & Van Riet I (2018). Mesenchymal stem cells in multiple myeloma: a therapeutical tool or target? Leukemia 32: 1500–1514. doi: 10.1038/s41375-018-0061-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z, Hou X, Lei W, Xiao L, Ebetino FH, Boeckman RK, et al. (2016). Bone targeted chloroquine inhibits osteoclastogenesis and bone resorption more effectively than chloroquine. J Bone Miner Res 31: S225. [Google Scholar]
- Young RN, & Grynpas MD (2018). Targeting therapeutics to bone by conjugation with bisphosphonates. Curr Opin Pharmacol 40: 87–94. doi: 10.1016/j.coph.2018.03.010 [DOI] [PubMed] [Google Scholar]
- Yun Z, Zhichao J, Hao Y, Ou J, Ran Y, Wen D, et al. (2017). Targeting autophagy in multiple myeloma. Leuk Res 59: 97–104. doi: 10.1016/j.leukres.2017.06.002 [DOI] [PubMed] [Google Scholar]
- Zangari M, & Suva LJ (2016). The effects of proteasome inhibitors on bone remodeling in multiple myeloma. Bone 86: 131–138. doi: 10.1016/j.bone.2016.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavrski I, Krebbel H, Wildemann B, Heider U, Kaiser M, Possinger K, et al. (2005). Proteasome inhibitors abrogate osteoclast differentiation and osteoclast function. Biochem Biophys Res Commun 333: 200–205. doi: 10.1016/j.bbrc.2005.05.098 [DOI] [PubMed] [Google Scholar]
- Zeng R, Chen Y, Zhao S, & Cui GH (2012). Autophagy counteracts apoptosis in human multiple myeloma cells exposed to oridonin in vitro via regulating intracellular ROS and SIRT1. Acta Pharmacol Sin 33: 91–100. doi: 10.1038/aps.2011.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, He J, Liu Z, Lu Y, Zheng Y, Li H, et al. (2015). Anti-beta(2)-microglobulin monoclonal antibodies overcome bortezomib resistance in multiple myeloma by inhibiting autophagy. Oncotarget 6: 8567–8578. doi: 10.18632/oncotarget.3251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Huo Q, Xu M, Yang F, Li Y, Shi H, et al. (2018). Bortezomib-catechol conjugated prodrug micelles: combining bone targeting and aryl boronate-based pH-responsive drug release for cancer bone-metastasis therapy. Nanoscale 10: 18387–18397. doi: 10.1039/c8nr03899f [DOI] [PubMed] [Google Scholar]
- Zinnen S, Rowinsky EK, Alexandrov A, Plekhova L, Roudas M, & Karpeisky A (2017). Phase 1 study of the bone-targeting cytotoxic conjugate, etidronate-cytosine arabinoside (MBC-11), in cancer patients with bone metastases. Journal of Clinical Oncology 35: 2589–2589. doi: 10.1200/JCO.2017.35.15_suppl.2589 [DOI] [Google Scholar]
- Zinnen SP, Karpeisky A, Von Hoff DD, Plekhova L, & Alexandrov A (2019). First-in-Human Phase I Study of MBC-11, a Novel Bone-Targeted Cytarabine-Etidronate Conjugate in Patients with Cancer-Induced Bone Disease. Oncologist 24: 303–e102. doi: 10.1634/theoncologist.2018-0707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, et al. (1998). CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev 12: 982–995. doi: 10.1101/gad.12.7.982 [DOI] [PMC free article] [PubMed] [Google Scholar]