

Abstract
Inflammasomes are intracellular protein complexes that drive the activation of inflammatory caspases1. To date, four inflammasomes involving NLRP1, NLRP3, NLRC4 and AIM2 have been described that recruit the common adaptor ASC to activate caspase-1, leading to the secretion of mature IL-1β and IL-182,3. The NLRP3 inflammasome has been implicated in the pathogenesis of several acquired inflammatory diseases4,5 as well as Cryopyrin-associated periodic fever syndromes (CAPS) caused by inherited NLRP3 mutations6,7. Potassium efflux is a common step that is essential for NLRP3 inflammasome activation induced by multiple stimuli8,9. Despite extensive investigation, the molecular mechanism leading to NLRP3 activation in response to potassium efflux remains unknown. We report here the identification of Nek7, a member of the family of mammalian NIMA-related kinases (Neks)10, as an NLRP3-binding protein that acts downstream of potassium efflux to regulate NLRP3 oligomerization and activation. In the absence of Nek7, caspase-1 activation and IL-1β release were abrogated in response to signals that activate NLRP3, but not NLRC4 or AIM2 inflammasome. NLRP3-activating stimuli promoted the NLRP3-Nek7 interaction in a process dependent on potassium efflux. NLRP3 associated with the catalytic domain of Nek7, but the catalytic activity of Nek7 was dispensable for activation of the NLRP3 inflammasome. Activated macrophages formed a high-molecular-mass NLRP3-Nek7 complex, which along with ASC oligomerization and ASC speck formation were abrogated in the absence of Nek7. Nek7 was required for macrophages harboring the CAPS-associated NLRP3R258W activating mutation to activate caspase-1. Mouse chimeras reconstituted with wild-type, Nek7−/− or Nlrp3−/− hematopoietic cells revealed that Nek7 was required for NLRP3 inflammasome activation in vivo. These studies demonstrate that Nek7 is an essential protein that acts downstream of potassium efflux to mediate NLRP3 inflammasome assembly and activation.
To understand the signaling mechanism of NLRP3 inflammasome activation, we sought to identify proteins that interact with NLRP3 upon inflammasome activation. To purify NLRP3 protein complexes, we generated a triple-tagged NLRP3 (NLRP3-SFP) fused with three tags in the carboxyl terminus: S-tag, FLAG (for detection), and a streptavidin-binding tag. Reconstitution of Nlrp3−/− immortalized bone-marrow-derived macrophages (iBMDMs) with NLRP3-SFP restored ATP-induced caspase-1 activation and IL-1β release (Extended Data Fig. 1a). We treated LPS-primed reconstituted iBMDMs with ATP to induce NLRP3 activation and searched for interacting partners of NLRP3 using liquid chromatography-mass spectrometry. The analysis revealed Nek7 as a major interacting partner of NLRP3 (Fig. 1a). NLRP3 did not associate with Nek6, a Nek7-related paralogue, or Nek9, another member of the Nek family10 (Extended Data Fig. 1b). The NLRP3-Nek7 interaction was confirmed by pull-down assays using streptavidin beads or immunoprecipitation (Fig. 1b, c and Extended Data Fig. 1b–d). Notably, the NLRP3-Nek7 interaction was slightly increased by LPS priming, but was clearly enhanced after ATP stimulation (Fig. 1a–c and Extended Data Fig. 1b–d). The interaction of NLRP3 with Nek7 was independent of ASC, caspase-1 or caspase-11 (Extended Data Fig. 1d). To determine the regions within NLRP3 that associate with Nek7, we expressed FLAG-tagged wild-type (WT) or mutant NLRP3 in HEK293T cells. Nek7 interacted with WT or mutant NLRP3 lacking the N-terminal Pyrin domain, but not NLRP3 with deletion of the C-terminal leucine-rich repeats (LRRs) (Fig. 1d). Furthermore, Nek7 did not associate with the singly expressed Pyrin domain, the centrally located nucleotide-binding domain (NOD) or LRRs (Fig. 1e), indicating that both the NOD and LRRs are involved in the interaction with Nek7. Conversely, the catalytic domain of Nek7, but not the N-terminal 33-amino acid extension domain interacted with NLRP3 (Fig. 1f). Further analysis showed that the N-terminal region (amino acid residues 34-212), but not the C-terminal region (residues 213-302) of the Nek7 catalytic domain11 mediates the interaction with NLRP3 (Fig. 1f). However, mutations of the catalytic K63 and K64 or the G43 amino acid residue that abolish the catalytic activity of Nek712,13 did not impair the NLRP3-Nek7 interaction (Fig. 1f). These results indicate that Nek7 interacts with NLRP3 and the interaction is enhanced in response to NLRP3-activating stimuli.
Figure 1. Nek7 interacts with NLRP3.

a, Mass spectrometry analysis of NLRP3 and Nek7 peptides after purification of NLRP3-associated proteins. b, NLRP3-SFP was pulled down and immunoblotted with indicated antibodies. c, LPS-primed BMDMs were left unstimulated or stimulated with ATP for 30 min. Cell lysates were immunoprecipitated and immunoblotted with indicated antibodies. d, e, WT or mutant NLRP3 was expressed in HEK 293T cells, immunoprecipitated and analysed by immunoblotting. f, WT or mutant Nek7 was co-expressed with FLAG-tagged NLRP3 in HEK 293T cells, immunoprecipitated and analysed by immunoblotting. Whole cell lysates are shown as the input. Results are representative of at least three independent experiments. See Supplementary Fig. 1 for gel source data.
We next evaluated the requirement for Nek7 in NLRP3 inflammasome activation. Because Nek7 deficiency leads to either embryonic lethality or death of pups soon after birth14, we generated mouse chimeras after transplanting fetal liver cells from Nek7+/+ or Nek7−/− embryos into lethally-irradiated recipient mice. BMDMs from mice reconstituted with Nek7−/− cells lacked detectable expression of Nek7, but expressed normal amounts of NLRP3, caspase-1, and ASC (Fig. 2a). Importantly, activation of caspase-1 and IL-1β release induced by ATP, nigericin and toxin gramicidin, three stimuli that activate NLRP3, were abolished in Nek7−/− BMDMs (Fig. 2b, c). In contrast, activation of caspase-1 and IL-1β release in response to poly(dA:dT) that activates the AIM2 inflammasome, or Salmonella enterica serovar Typhimurium (Salmonella) that activates the NLRC4 inflammasome, were not affected in Nek7−/− BMDMs (Fig. 2b, c). Likewise, caspase-1 activation and IL-1β release induced by particulate matter and the lysosome membrane damaging agent Leu-Leu-OMe (LLOMe), were impaired in Nek7−/− BMDMs (Fig. 2d, e). In contrast, TNF-α release induced by all tested stimuli was unaffected in Nek7−/− BMDMs (Extended Data Fig. 2a, b). In addition, NLRP3-dependent caspase-1 activation and IL-1β release induced by cytosolic LPS stimulation that activates the non-canonical inflammasome via caspase-11 also required Nek7 (Extended Data Fig. 2c, d). Consistent with previous studies15–17, cytotoxicity induced by cytosolic LPS required caspase 11, but not NLRP3 or Nek7 (Extended Data Fig. 2e). To ensure that impaired NLRP3 activation in Nek7−/− BMDMs was not secondary to abnormal mouse development, we deleted Nek7 using CRISPR/Cas9 genome editing in iBMDMs. NLRP3 inflammasome activation induced by nigericin was abrogated in Nek7-deficient macrophages (Fig. 2f, g and Extended Data Fig. 3a–c). Importantly, re-expression of Nek7 in Nek7-deficient macrophages restored NLRP3 inflammasome activation (Fig. 2f, g). Likewise, knockdown of Nek7 by short hairpin RNAs targeting Nek7 impaired caspase-1 activation and IL-1β release, but not TNF-α production, in response to ATP, nigericin or silica (Extended Data Fig. 4a–d). We also depleted Nek7 in BMDMs harboring the activating Nlrp3R258W mutation corresponding to the human NLRP3R260W mutation that causes Muckle-Wells syndrome18. In agreement with previous studies19, treatment of Nlrp3R258W BMDMs with LPS alone was sufficient to activate caspase-1 and IL-1β release (Fig. 2h, i). Notably, caspase-1 activation and IL-1β release elicited by LPS in Nlrp3R258W BMDMs were impaired by Nek7 knockdown (Fig. 2h, i). These results indicate that Nek7 acts on or just downstream of both WT and CAPS-associated NLRP3 to regulate the inflammasome.
Figure 2. Nek7 deficiency specifically abrogates the activation of the NLRP3 inflammasome.

a, BMDMs were left untreated or stimulated with LPS and cell lysates were immunoblotted with indicated antibodies. b, d, Caspase-1 in the supernatant (Sup.) and cell lysate (Lys.) was analysed in stimulated Nek7+/+ and Nek7−/− macrophages. c, e, IL-1β release was measured. f, g, CRISPR/Cas9-generated Nek7-deficient iBMDMs were transduced with control or pHIV-Nek7 lentivirus, and stimulated with LPS plus nigericin. Caspase-1 activation (f) and IL-1β release (g) were analysed. h, i, Macrophages were transduced with control lentivirus or shRNA lentiviruses targeting Nek7, and stimulated with LPS. Caspase-1 activation (h) and IL-1β release (i) were analysed. IL-1β release data (c, e, g, i) are expressed as mean values. Error bars indicate ± s.d. of triplicate wells. Results are representative of three independent experiments. See Supplementary Fig. 1 for gel source data.
Stimulation of Nek7+/+ BMDMs with the NLRP3 activators ATP and nigericin, as well as poly(dA:dT) or Salmonella, induced rapid formation of large intracellular ASC aggregates called ASC specks in the cytosol (Fig. 3a, b). The formation of ASC specks induced by ATP or nigericin was abrogated, but unperturbed when induced by poly(dA:dT) or Salmonella, in Nek7−/− BMDMs (Fig. 3a, b). Consistently, ASC oligomerization triggered by stimulation with multiple NLRP3 activators, but not poly (dA:dT) or Salmonella infection, was abolished in Nek7−/− BMDMs or greatly reduced in BMDMs with knockdown of Nek7 (Fig. 3c and Extended Data Fig. 5a–c). Activated inflammasomes assemble into high-molecular-mass multiprotein complexes1,20. To assess NLRP3 inflammasome assembly, WT and Nek7−/− BMDMs were stimulated with nigericin or ATP, and digitonin-solubilized cell lysates were resolved by blue native polyacrylamide gel electrophoresis (PAGE) and then the blots were immunoblotted with anti-NLRP3 and anti-Nek7 antibodies. A large oligomeric complex (> 1,000_kDa) containing NLRP3 and Nek7 was induced in WT BMDMs after stimulation with ATP or nigericin which was greatly reduced or absent in stimulated Nek7−/− and Nlrp3−/− cells (Fig. 3d ). To better resolve the formation of NLRP3 oligomers, we separated the samples in the first dimension by blue native PAGE and then in a second dimension by SDS–PAGE. Immunoblotting revealed that Nek7 was indeed present in a high-molecular-mass NLRP3 complex induced by ATP or nigericin in primary WT BMDMs, which was absent in unstimulated WT cells and stimulated Nek7−/− or Nlrp3−/− cells (Fig. 3e). To determine whether the kinase activity of Nek7 is important for the regulation of the NLRP3 inflammasome, we expressed WT or mutant Nek7 with mutations at the critical catalytic lysine residues K63 and K64 or glycine residue G43 in Nek7−/− iBMDMs by lentivirus-mediated transduction. All of these Nek7 mutations have been found to abolish the kinase activity of Nek712,13. Importantly, the ability of nigericin to induce caspase-1 activation and IL-1β release was not impaired in Nek7−/− iBMDMs reconstituted with these Nek7 mutant proteins (Extended Data Fig. 6a–c). In addition, depletion of Nek9, the kinase that phosphorylates and activates Nek711,21 or the Nek7-related kinase Nek6 by shRNAs, did not impair the activation of the NLRP3 inflammasome (Extended Data Fig. 7a–e). Nek7 regulates microtubule dynamics22,23. However, treatment with nocodazole or colchicine, two inhibitors of tubulin polymerization, did not inhibit NLRP3 inflammasome activation and had a marginal inhibitory effect on IL-1β and TNF-α release (Extended Data Fig. 8a–e). Furthermore, inhibition of tubulin polymerization did not affect the NLRP3-Nek7 interaction (Extended Data Fig. 8f). Inhibition of the Bruton’s tyrosine kinase (BTK) that has been implicated in NLRP3 inflammasome activation24 did not disrupt the interaction between Nek7 and NLRP3 (Extended Data Fig. 9a, b). Additionally, we could not detect BTK in NLRP3 complexes before or after ATP stimulation (Extended Data Fig. 9b). Stimulation of Nek7+/+ and Nek7−/− BMDMs with ATP, nigericin or gramicidin induced comparable potassium efflux (Extended Data Fig. 10). Notably, the association of NLRP3 with Nek7 as well as caspase-1 activation triggered by ATP were blocked in the presence of 50 mM KCl that inhibits potassium efflux and NLRP3 activation (Fig. 3f). Importantly, the induced high-molecular-mass NLRP3 complex containing Nek7 was not seen in BMDMs cultured in 50 mM KCl (Fig. 3g). These results indicate that potassium efflux promotes the NLRP3-Nek7 interaction that is required for NLRP3 inflammasome assembly and caspase-1 activation (Fig. 3h).
Figure 3. Nek7 is required for NLRP3 oligomerization and ASC speck formation downstream of potassium efflux.

a, b, Representative immunofluorescence images and quantification of endogenous ASC specks (arrows). N.D. not detected. Data shown represent results from three combined independent experiments. Scale bars, 10 μm. Error bars indicate ± s.d. c, ASC oligomerization induced by indicated stimuli in Nek7+/+ and Nek7−/− LPS-primed macrophages. d, Indicated LPS-primed macrophages were stimulated with PBS (Mock), ATP or nigericin. Analyses by blue native PAGE or SDS-PAGE, and immunoblotting. e, Cell lysates were separated by a first dimension of blue native PAGE followed by a second dimension of SDS–PAGE. f, g, Nek7/NLRP3 interactions and complex formation in the presence of 5 mM or 50 mM extracellular KCl. h, Proposed model for mechanism of Nek7-mediated NLRP3 inflammasome activation. Results are representative of at least three independent experiments. See Supplementary Fig. 2 for gel source data.
We assessed the role of Nek7 in the regulation of the inflammasome in vivo. We generated mouse chimeras after transplanting fetal liver cells from WT, Nek7−/− or Nlrp3−/− embryos into lethally-irradiated recipient mice, and IL-1β production induced by LPS challenge was assessed in the serum25,26. Administration of LPS to chimeric mice reconstituted with WT fetal liver cells induced IL-1β production, which was diminished similarly in chimeric mice reconstituted with Nek7−/− or Nlrp3−/− hematopoietic cells (Fig. 4a). Consistent with additional NLRP3 function in radio-resistant recipient cells27, chimeric Nlrp3−/− recipients transplanted with Nek−/− or Nlrp3−/− fetal liver cells showed further reduction in IL-1β production (Fig. 4a). Importantly, production of IL-6 and TNF-α in the sera of all chimeric mice was comparable (Fig. 4b, c). Likewise, production of IL-1β induced by intraperitoneal administration of MSU crystals that is also largely mediated by the NLRP3 inflammasome28, was reduced at comparable levels in chimeric mice reconstituted with Nek7−/− or Nlrp3−/− fetal liver cells (Fig. 4d). These studies indicate that Nek7 is required for activation of the NLRP3 inflammasome in vivo.
Figure 4. Nek7 is required for activation of the NLRP3 inflammasome in vivo.

a–c, Mouse serum cytokines IL-1β (a), TNF-α (b) and IL-6 (c) were analysed after intraperitoneal injection of LPS (LPS i.p.). d, IL-1β was analysed in peritoneal lavage fluids after intraperitoneal injection of MSU (MSU i.p.). Each symbol represents one mouse. Mean values are indicated by a horizontal bar. In the absence of LPS or MSU stimulation, the amounts of IL-1β were undetectable. Results are representative of two independent experiments. ns, not significant. *p<0.05; **, p<0.01; *** p<0.001 by Kruskal-Wallis test.
We have shown that Nek7 is an essential factor that specifically and non-redundantly functions downstream of potassium efflux to regulate the activation of the NLRP3 inflammasome. Our results are in agreement with recent studies that identified Nek7 as a critical regulator of the NLRP3 inflammasome29,30. Our studies suggest a model in which potassium efflux induced by NLRP3-activating stimuli triggers the association of NLRP3 with Nek7, leading to the assembly and activation of the NLRP3 inflammasome (Fig. 3h). Macrophages harboring the CAPS-associated Nlrp3R258W activating mutation that does not require potassium efflux for inflammasome activation19 also required Nek7 for caspase-1 activation, suggesting that this mutant NLRP3 may be competent for Nek7 association in the absence of potassium efflux. Nek7 regulates microtubule dynamic instability and spindle assembly which required the catalytic activity of Nek712,13,22,23. In contrast, the catalytic activity of Nek7 is not required for NLRP3 activation. Further work is needed to understand the dual functions of Nek7. Taken together, our studies suggest that Nek7 could be a potential target of therapeutics to treat inflammatory diseases linked to NLRP3 inflammasome activation.
Methods
Mice
Nek7+/−, Nlrp3−/−, Asc−/−, Casp1−/−Casp11−/−, Casp11−/− mice on C57BL/6 background have been reported14,31–33. Nlrp3R258W mice were originally provided by Warren Strober (NIH). C57BL/6 mice were originally purchased from Jackson Laboratories (Bar Harbor, ME, USA) and maintained in our facility. All animal studies were approved by the University of Michigan Committee on Use and Care of Animals.
Reagents
High Capacity streptavidin agarose resin was from Thermo Scientific (20359). S-protein agarose beads were from Novagen (69704). Biotin (B4501), nocodazole (M1404) and colchicine (C3915) were purchased from Sigma. LFM-A13 (1300) was purchased from Tocris. DOTAP liposomal transfection reagent was from Roche (11202375001). CytoTox 96 Non-Radioactive Cytotoxicity Assay Kit was purchased from Promega (G1780). LPS-B5 Ultrapure (tlrl-pb5lps), LPS-SM Ultrapure (tlrl-smlps), MSU (tlrl-msu), CPPD (tlrl-cppd), Nano-SiO2 (tlrl-sio) and poly(dA:dT)/lyovec (tlrl-patc) were purchased from InvivoGen. Alum (77161) was from Thermo Fisher Scientific. ATP (A2383) was from Sigma. Nigericin (481990) was purchased from EMD Millipore. Silica (Min-U-Sil 5) was from US Silica. LLOMe was purchased from Chem-Impex. Salmonella enterica serovar Typhimurium strain SL1344 was a gift of Denise Monack (Stanford University). Human Nek7 cDNA clone was purchased from the Harvard DNA Resource Core (HsCD00082815). Mouse Nek7 cDNA clone was from Thermo Scientific (MMM1013-202764053). Lentiviral expression vector pHIV-EGFP was from Adgene (21373). QuikChange II XL Site-Directed Mutagenesis Kit was from Agilent (20051). Antibodies for Nek7 (ab133514), Nek6 (ab133494), Nek9 (ab138488) were purchased from Abcam. Mouse anti-Nlrp3 (Cryo-2) was from AdipoGen2. Actin (A00730-200), HA tag (A01244-100) and FLAG (DYKDDDDK) tag (A00187-200) antibodies were purchased from GenScript. Anti-α/β tubulin and anti-Btk antibodies were from Cell signaling (2148, 3533). ASC antibody and Caspase-1 antibody for the cleaved p20 of Caspase-1 were generated in our laboratory.
Cell culture
Bone marrow derived macrophages (BMDMs) were generated by differentiating bone marrow progenitors from the tibia and femur for 7 days in 10% FCS IMDM (Gibco) supplemented with 30% L-cell supernatant, non-essential amino acids, sodium pyruvate and antibiotics (Penicillin/Streptomycin). Immortalized bone marrow derived macrophages (iBMDMs) were generated as previously described 34. iBMDMs were cultured in 10% FCS IMDM (Gibco) supplemented with non-essential amino acids, sodium pyruvate and antibiotics (Penicillin/Streptomycin). HEK 293T cells (ATCC) were cultured on Dulbecco’s Modified Eagle’s medium (Sigma) containing 10% FCS and antibiotics (Penicillin/Streptomycin).
Purification of NLRP3-associated proteins
To search for interacting partners of NLRP3 in mouse macrophages, we generated a triple-tagged NLRP3 (Nlrp3-SFP) that was fused with three tags in the carboxyl terminus: S-tag, FLAG (for detection), and streptavidin-binding tag. This tagged NLRP3 can be used to purify NLRP3 protein without antibody-based immunoprecipitation. NLRP3-SFP was cloned into the pHIV-EGFP lentiviral expression vector (Addgene). Transduced immortalized Nlrp3−/− macrophage clones were sorted by flow cytometry using GFP as a marker and screened for NLRP3 expression by immunoblotting. Clones with NLRP3 expression levels comparable to those found in wild-type iBMDMs stimulated with LPS were selected for further analysis. Macrophages were plated in 10-cm Petri dishes and primed with 200 ng/ml LPS for 4 h before stimulation with 5 mM ATP for 30 min. Cells were washed once with cold PBS after stimulation and then lysed in ice-cold lysis buffer (50 mM Tris-HCl, pH7.4, 2 mM EDTA, 150 mM NaCl, 0.5% Nonidet P-40, 1x EDTA-free Roche protease inhibitor cocktail) for 10 min at 4 °C. Cell lysates were collected and spun down at 20,000 × g for 15 min at 4 °C. The soluble fraction was incubated with streptavidin agarose beads for 2 h at 4 °C. The beads were washed three times with lysis buffer. Bound proteins were eluted with 2 mM biotin in PBS, and then re-captured by incubation with S-protein agarose beads. The NLRP3-associated proteins were finally eluted in SDS sample buffer (10% glycerol, 60 mM Tris-HCl, pH6.8, 2% SDS, 0.02% bromophenol blue and 5% β-mercaptoethanol), separated by 4–12 % SDS–PAGE, followed by mass spectrometry. Nlrp3−/− macrophages reconstituted with empty vector (pHIV-EGFP) were used as a control.
HEK293T Cell Transfection
HEK 293T cells were plated into 6-well tissue culture plates (6.25 x10^5 cells per well in 2 ml complete DMEM) overnight. Cells were transfected for 16 h with triple FLAG-tagged full-length NLRP3 (FL, 0.5 μg), Pyrin domain deletion mutant ΔPyrin (amino acids 94–1036, 0.5 μg), LRR deletion mutant ΔLRR (amino acids 1–741, 1 μg), Pyrin domain (amino acids 1–93, 0.5 μg), NOD domain (amino acids 220–536, 2.5 μg), LRR domain (amino acids 742–991, 1 μg), or co-transfected FLAG- tagged full-length NLRP3 (0.5 μg) with Nek7-3xHA (0.5 μg) by Lipofectamine LTX (Invitrogen).
Immunoprecipitation
Cells were lysed in ice-cold lysis buffer (50 mM Tris, pH 7.4, 2 mM EDTA, 150 mM NaCl, 0.5% Nonidet P-40, 1x EDTA-free Roche protease inhibitor cocktail). Cell lysates were clarified by centrifugation (14,000 × g) at 4 °C for 10 min. Pre-cleared cell lysates were incubated with anti-Nek7 (1:100), anti-Nlrp3 (1:100), anti-FLAG (1:200), anti-HA (1:200) antibody or control IgG at 4 °C overnight. The proteins bound by antibody were pulled down by protein G beads and subjected to immunoblotting analysis.
Inflammasome activation and cytotoxicity assay
Macrophages were plated in 12-well plates at 1x106 cells per well. One day later, culture medium was replaced with 0.5 ml serum-free IMDM per well. Cells were then primed with 200 ng/ml ultrapure LPS for 4 h, followed by stimulation with PBS (mock), 5 mM ATP (30 min), 5 μM nigericin (1 h), 0.5 μM gramicidin (1 h), poly(dA:dT) (2 μg/ml, 4 h), Salmonella (m.o.i=10, 1h), LLOMe (2 μM, 6h), Silica (500 μg/ml, 6h), Alum (250 μg/ml, 6h), MSU (200 μg/ml), CPPD (200 μg/ml, 6h) or Nano-SIO2 (200 μg/ml, 6h). After stimulation, supernatants and cell lysates were collected separately, or combined together for immunoblotting analysis. For non-canonical inflammasome activation, 1 μg of LPS-SM was packaged with 15 μl of DOTAP according to manufacturer’s instructions. After 30 min incubation at room temperature, the mixtures were diluted into 1 ml Opti-MEM and added to each well of 6-well plate containing (LPS-B5)-primed macrophages (4 h priming). 4 h later, culture supernatants and cell lysates were collected for immunoblotting. 48-well plates were used for IL-1β release and cytotoxicity assay.
Gene knockdown in primary macrophages
lentiviral pLKO.1 plasmids targeting Nek7, Nek6 and Nek9 were purchased from Sigma. Nek7 shRNA1 (TRCTRCN0000236005, targeting sequence: CCGGTGGAGTGCCGGTAGCGTTAAACTCGAGTTTAACGCTACCGGCACTCCATTTTTG), Nek7 shRNA2 (TRCN0000236007, CCGGGAGCTACGACAGCTAGTTAATCTCGAGATTAACTAGCTGTCGTAGCTCTTTTTG), Nek6 shRNA(TRCN0000274723, CCGGAGGACAGTTCAGTGAGGTTTACTCGAGTAAACCTCACTGAACTGTCCTTTTTTG) and Nek9 (TRCN0000027597, CCGGCGACAACATCATTGCCTACTACTCGAGTAGTAGGCAATGATGTTGTCGTTTTT) were reported in this study. pLKO.1 scramble (Addgene ID#1864) was used as a negative control. Lentiviruses were packaged in HEK293FT cells and concentrated by Lenti-X concentrator (Clontech). Gene knockdown in primary macrophages was performed as described previously20. Puromycin-selected macrophages were collected, counted and plated on day 8, and used for experiments on the next day. The knockdown efficiency was analysed by immunoblotting with antibodies against Nek7, Nek6 or Nek9.
Targeting of Nek7 by CRISPR/Cas9-mediated genome editing
Lentiviral CRISPR/Cas9 targeting guide RNA (gRNA) expressing vector (lentiCRISPRv2) was obtained from Addgene (#52961). The Nek7 knockout (KO) target sequence used was 5′-TGAAAAACCGACCAAGCCCA-3′. To generate a Nek7 KO cell line, lentiCRISPRv2 stocks containing the target sequence 5′-TGAAAAACCGACCAAGCCCA-3′ were used to transduce iBMDMs. 3 days later, single clones of puromycin-resistant cells were sorted by flow cytometry and clones with Nek7-deficiency were identified by immunoblotting with anti-Nek7 antibody.
Reconstitution of Nek7 in Nek7−/− macrophages
Immortalized Nek7−/− macrophages were transduced with lentivirus containing pHIV-Nek7-3xHA. After 3–4 days, transduced cells were sorted by flow cytometry using EGFP as a marker. Expression of reconstituted proteins was determined by immunoblotting.
Generation of chimeras from fetal liver cells
Fetal livers were harvested at day 14 of gestation from WT, Nek7−/− or Nlrp3−/− embryos. WT and Nek7−/− fetuses were genotyped with primers (WT, forward, 5′-AAGGCTTACTTGGTGACACACTGG-3′; reverse, 5′-CACCGTGCAGGTGACTCGAACC-3′; Nek7−/−, forward, 5′′-CCCTGGCGTTACCCAACTTAATCGCCTTGC-3′, reverse, 5′-TTCTCCGTGGGAACAAACGGCGGATTGAC-3′), which yielded a 400-base-pair (bp) wild-type DNA band and a 302-bp Nek7 mutant DNA band after electrophoresis on 1.5% agarose gel in TBE buffer. Fetal livers were harvested from fetuses and processed into single cell suspension using a 40 μm Nylon Cell Strainer (Falcon). 5 × 106 fetal liver cells in 100 μl PBS were administered by retro-orbital injection into 6-week-old gender-matched WT or Nlrp3−/− recipient mice that had been lethally irradiated using X-ray (Phillips RT250, Kimtron Medical) with two doses of 540 rad (total 1080). Recipient mice were allocated randomly into experimental groups. Recipient animals received antibiotics (Neomycin and Polymycin B) for 4 weeks after reconstitution. 8 weeks later, successful generation of chimeras was confirmed by PCR-based analysis and animals were analysed (see above). The number of animals per group (n= 5–8) was chosen as the minimum likely required for conclusions of biological significance, established from prior experience.
ASC speck staining and ASC oligomer cross-linking
BMDMs were plated on an 8-well permanox chamber slide (Thermo scientific, cat no. 177445) overnight. Cells were primed with 200 ng/ml LPS for 4 h, then stimulated with ATP, nigericin, transfected with poly(dA·dT), or infected with Salmonella. After stimulation, cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and the slides blocked with PBS buffer containing 3% BSA. Cells were stained with anti-ASC antibody and Alexa Fluor 488-conjugated secondary antibody. DAPI was used to stain nuclei. Cell images were taken using an Olympus Fluo-View 500 confocal microscope system.
For ASC oligomer cross-linking, cells were plated on 12-well plates and stimulated as indicated. Cells were lysed with PBS buffer containing 0.5% Triton X-100, and the cell lysates were centrifuged at 8000 rpm for 15 min at 4 °C. Supernatants were transferred to new tubes (TX-soluble fractions). The Triton X-100-insoluble pellets were washed with PBS twice and then suspended in 200 μl of PBS. The pellets were then cross-linked at room temperature for 30 min by adding 2 mM bis[sulfosuccinimidy] suberate (BS3). The cross-linked pellets were spun down at 8000 rpm for 15 min and dissolved directly in SDS sample buffer.
Blue native PAGE and 2D PAGE
Blue native gel electrophoresis was performed using the Bis-Tris Native PAGE system as previously described20,35. Briefly, 2 x106 macrophages were re-plated in 6-well plates. On the second day, cells were primed with 200 ng/ml LPS for 4 h, followed by stimulation with PBS (mock), 5 mM ATP (30 min) or 5 μM nigericin (1h). Macrophages were washed once with cold PBS and then lysed in ice-cold native lysis buffer (20 mM Bis-tris, 500 mM ε-aminocaproic acid, 20 mM NaCl, 10% w/v glycerol, 0.5% digitonin, 0.5 mM Na3VO4, 1mM PMSF, 0.5 mM NaF, 1x EDTA-free Roche protease inhibitor cocktail, pH7.0) for 15 min on ice. Cell lysates were clarified by centrifugation at 20,000g for 30 min at 4 °C and analysed without further purification steps. Cell lysates were equalized after quantification of total protein using the BCA protein assay (Pierce), and then separated by 4–12 % blue native PAGE. Native gels were incubated in 10% SDS solution for 5 min before transfer to PVDF membranes (Millipore), followed by conventional Western blotting. In 2D PAGE, a gel slice of the natively resolved gel was placed in a dish containing 1x Laemmli sample buffer (12.5mM Tris-Cl (pH 6.8), 5% β-mercaptoethanol4% (w/v) SDS, 0.1% bromophenol blue, 20% (v/v) glycerol) for 10 min, microwaved on high for 20 s, and rocked for another 15 min before loading the slice into a well 4–12% SDS–PAGE gel. Because the speck was mostly lost in the pellet fraction, the analyses of the Nek7-NLRP3 interaction likely have excluded proteins in the speck.
Potassium efflux assay
Intracellular potassium measurements were performed as described previously8. Briefly, macrophages were plated on 96-well plates 1 day before experiments. Culture medium was thoroughly aspirated after stimulation. Cells were lysed with 3% ultrapure HNO3. Intracellular potassium was then determined by inductively coupled plasma optical emission spectrometry with an Optima 2000 DV spectrometer (PerkinElmer Life Sciences) using yttrium as an internal standard.
Cytokine measurements
Cytokines were measured with enzyme-linked immunoabsorbent assay (ELISA) kits (R&D Systems).
Stimulation with endotoxin in vivo
Mice were injected intraperitoneally with 20mg/kg LPS (E. coli 0111:B4; Sigma). The investigators were not blinded to allocation during experiments. The blood samples were collected 3 h later. Serum cytokines were measured by ELISA. The analysis was performed blindly by an independent researcher.
MSU-induced mouse peritonitis
Chimeric mice were injected intraperitoneally with 1 mg MSU dissolved in 0.5 ml sterile PBS. Mice were sacrificed 6 h later and peritoneal cavities were flushed with 5 ml cold PBS. Peritoneal lavage fluids were collected and cytokines were measured by ELISA after concentration using an Amicon Ultra 10K filter from Millipore. The investigators were not blinded to allocation during experiments. Serum cytokines were measured by ELISA. The analysis was performed blindly by an independent researcher.
Statistical Analysis
All analyses were performed using GraphPad Prism. Differences were considered significant when p values were less than 0.05.
Extended Data
Extended Data Figure 1. Nek7 interacts with NLRP3 upstream of ASC and Caspase-1.

a, Immortalized Nlrp3−/− macrophages were infected with lentiviruses containing pHIV vector (Vector) or pHIV-NLRP3-SFP (NLRP3-SFP). Transduced macrophages were sorted by flow cytometry. Cells were treated as indicated. Supernatants (Sup.) and cell lysates (Lys.) were collected and analysed by immunoblotting with indicated antibodies. b, Immunoblots showing that Nek7, but not Nek6 or Nek9, interacts with NLRP3 in macrophages. NLRP3-associated proteins were pulled down from reconstituted Nlrp3−/− macrophages by streptavidin beads, and analysed by immunoblotting with indicated antibodies. c, ATP stimulation, but not LPS priming, greatly enhances the Nek7/NLRP3 interaction. d, LPS-primed BMDMs from WT, Asc−/− and Casp1−/−/Casp11−/− were left untreated or treated with 5 mM ATP for 30 min. NLRP3 protein complexes were immunoprecipitated with anti-NLRP3 antibody and analysed by immunoblotting. Data are representative of three (a, b) or two (c, d) independent experiments. See Supplementary Fig. 2, 3 for gel source data.
Extended Data Figure 2. Nek7 deficiency does not affect TNF-α release, but abrogates NLRP3 activation induced by cytosolic LPS in macrophages.

a, TNF-α secretion from LPS-primed BMDMs treated with ATP, Nigericin, Gramicidin, Poly(dA:dT) or Salmonella. b, TNF-α secretion in LPS-primed BMDMs treated with LLOMe, Silica, Alum, MSU, CPPD or Nano-SiO2. Mock represents macrophages primed with LPS without further stimulations. The amounts of TNF-α release in the absence of LPS priming were below 50 pg/ml in the supernatants. c-e, BMDMs from C57BL/6, Casp11−/−, Nek7−/− (fetal liver chimeras) and Nlrp3−/− mice were primed with LPS for 4 h before transfection with or without LPS with DOTAP. 4 h after transfection, caspase-1 activation (c), IL-1β release (d) and cytotoxicity (e) were analysed. Graphs show the mean ± s.d. of triplicate wells and are representative of three independent experiments. See Supplementary Fig. 3 for gel source data.
Extended Data Figure 3. Nek7 is required for activation of the NLRP3 inflammasome in macrophages.

a–c, Defective NLRP3 inflammasome in Nek7-deficient macrophages. WT and Nek7-deficient macrophages generated by CRISPR/Cas9 genome editing were primed with LPS, followed by stimulation with 5 μM nigericin (1 h), 5 mM ATP (30 min) or 200 ng/ml Alum (6 h). Cell lysates were collected and analysed for caspase-1 activation by immunoblotting (a). IL-1β (b) and TNF-α (c) release were analysed by ELISA. ELISA data (b, c) show the mean ± s.d. of triplicate wells. The amounts of TNF-α release in the absence of LPS priming were below 50 pg/ml in the supernatants. Data are representative of three independent experiments. See Supplementary Fig. 3 for gel source data.
Extended Data Figure 4. Nek7 is critical for activation of the NLRP3 inflammasome in primary macrophages.

a, Nek7 is dispensable for NLRP3 induction and expression of the NLRP3 inflammasome components ASC and caspase-1. BMDMs were treated with control shRNA or Nek7 shRNAs and selected by culture in 3 μg/ml puromycin. Puromycin-resistant macrophages were left unstimulated or stimulated with LPS for 4 h. Cell lysates were collected and analysed by immunoblotting. Actin is shown as a loading control. b–c, Nek7 depletion inhibits activation of the NLRP3 inflammasome. BMDMs treated with control shRNA or Nek7 shRNAs were primed with LPS, followed by stimulation with 5 mM ATP (30 min), 5 μM nigericin (1 h) or 500 ng/ml silica (4 h). Supernatants (Sup.) and cell lysates (Lys.) were collected after stimulation and analysed by immunoblotting for caspase-1 activation (b). IL-1β (c) and TNF-α (d) release were analysed by ELISA. ELISA data (c, d) show the mean ± s.d. of triplicate wells. The amounts of TNF-α release in the absence of LPS priming were below 50 pg/ml in the supernatants. Data are representative of three independent experiments. See Supplementary Fig. 3 for gel source data.
Extended Data Figure 5. Nek7 is critical for NLRP3-mediated ASC oligomerization and speck formation in macrophages.

a, Representative confocal immunofluorescence images and (b) quantification of endogenous ASC specks (arrows) in macrophages treated with control shRNA- or Nek7 shRNA. Macrophages were primed with LPS and then stimulated with ATP, nigericin, Poly(dA:dT) or Salmonella. After stimulation, cells were fixed and stained for ASC (Green). DAPI was used for staining nuclear DNA (Blue). Data shown represent results from three combined independent experiments in which more than 300 cells were counted in each experiment. Error bars indicate ± s.d. c, Immunoblots showing ASC oligomerization in control shRNA or Nek7 shRNA-treated macrophages. LPS-primed macrophages were treated as indicated. Cell lysates (Triton-X-100 soluble) and BS3-crosslinked pellets (Triton-X-100 insoluble) were analysed by immunoblotting. Mock represents macrophages primed with LPS without further stimulations. Results are representative of two independent experiments. Two-tailed Student’s t-test. * p <0.05. See Supplementary Fig. 3 for gel source data.
Extended Data Figure 6. Nek7 kinase activity is dispensable for activation of the NLRP3 inflammasome in macrophages.

a–c, Immortalized Nek7-deficient macrophages were infected with lentivirus containing empty vector or vector expressing wild type or indicated mutant Nek7. Transduced macrophages were sorted by flow cytometry using GFP as a marker. Macrophages were plated and stimulated with 5 μM nigericin for 1 h after LPS priming. Cell lysates were collected and analysed for caspase-1 activation by immunoblotting (a). IL-1β (b) and TNF-α (c) release were analysed by ELISA. ELISA data (b, c) show the mean ± s.d. of triplicate wells. Data are representative of three independent experiments. See Supplementary Fig. 3 for gel source data.
Extended Data Figure 7. Nek6 and Nek9 are dispensable for activation of the NLRP3 inflammasome in macrophages.

a–c, BMDMs were treated with control shRNA, Nek7 shRNA, Nek6 shRNA or Nek9 shRNA and selected by culture with 3 μg/ml puromycin. Puromycin-resistant cells were plated and primed with LPS, followed by stimulation with 5 mM ATP for 30 min. Cell lysates were collected and analysed for caspase-1 activation by immunoblotting (a). IL-1β (b) and TNF-α (c) release were analysed by ELISA. d, Representative confocal immunofluorescence images and (e) quantification of endogenous ASC specks (arrows) in macrophages. DAPI was used for staining nuclear DNA (Blue). Quantification of ASC speck was from three combined independent experiments in which more than 300 cells were counted in each experiment. Error bars indicate ± s.d. ELISA data (b, c) show the mean ± s.d. of triplicate wells. Data are representative of three independent experiments. Two-tailed Student’s t-test. * p <0.05. See Supplementary Fig. 3 for gel source data.
Extended Data Figure 8. Microtubule depolymerization does not inhibit the activation of the NLRP3 inflammasome and the Nek7/NLRP3 interaction in macrophages.

a–e, LPS-primed BMDMs were pretreated with vehicle control (DMSO), nocodazole (1, 10, 50 μM) or colchicine (1, 10, 50 μM) for 1 h before stimulation with 5 μM nigericin. Cell lysates were collected and analysed for caspase-1 activation by immunoblotting (a). IL-1β (b) and TNF-α (c) release were analysed by ELISA. Representative confocal immunofluorescence images (d) and (e) quantification of endogenous ASC specks (arrows) in macrophages treated as indicated. Microtubules were stained with anti-α/β tubulin antibody (Red). DAPI was used for staining nuclear DNA (Blue). Data shown in (e) represent results from three combined independent experiments in which more than 300 cells were counted in each experiment. Error bars indicate ± s.d. f, The Nek7/NLRP3 interaction in untreated and treated macrophages was analysed by immunoprecipitation and immunoblotting. ELISA data (b, c) show the mean ± s.d. of triplicate wells. Data are representative of three independent experiments. Two-tailed Student’s t-test. * p <0.05. See Supplementary Fig. 3 for gel source data.
Extended Data Figure 9. Inhibition of Bruton’s tyrosine kinase (BTK) does not inhibit the Nek7/NLRP3 interaction in macrophages.

a–b, Reconstituted Immortalized Nlrp3−/− macrophages were primed with LPS for 4 h. Cells were left untreated or treated with indicated concentrations of BTK inhibitor LFM-A13 for 30 min before ATP stimulation. Cell lysates were collected and analysed for caspase-1 activation by immunoblotting (a). Cell lysates were collected and subjected to pull-down with streptavidin beads, and the precipitated protein complex was analysed by immunoblotting with indicated antibodies (b). Agarose beads were used as a control. Actin is shown as a loading control. Data are representative of two independent experiments. See Supplementary Fig. 3 for gel source data.
Extended Data Figure 10. Nek7-deficiency does not inhibit potassium efflux induced by NLRP3 stimuli in macrophages.
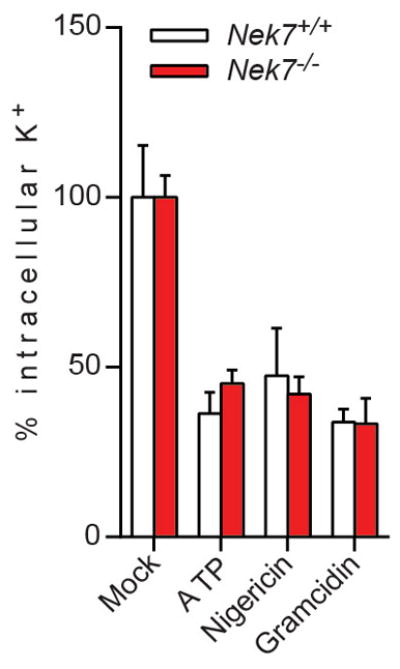
LPS-primed Nek7+/+ and Nek7−/− BMDMs were stimulated as indicated. The intracellular potassium in each condition was analysed. Mock represents macrophages primed with LPS without further stimulation. Graphs show the mean ± s.d. of four technical replicates and are representative of two independent experiments.
Supplementary Material
Acknowledgments
We thank R. Muñoz-Planillo for potassium efflux assays, S. Varadarajan for plasmid construction, L. Burmeister for animal husbandry, J. Whitfield for ELISA assays, and V. Basrur for mass spectrometry analysis. Y. H. was supported by NIH training grant T32HL007517. M.Y.Z. was supported by NIH training grants T32HL007517 and T32DK094775. D. Y. was supported by the State Scholarship Fund from China Scholarship Council (No. 201306740018). This work was supported by NIH grants R01AI063331 and R01DK091191 to G.N., research funds by the Israel Science Foundation (grant no. 768/11) to B. M., and funds to the Michigan Comprehensive Cancer Center Immunology Monitoring Core from the University of Michigan’s Cancer Center Support Grant.
Footnotes
Supplementary Information is available in the online version of the paper
Author Contributions Y. H. and G.N. designed the research and wrote the manuscript. Y. H. conducted the experiments and analysed data with the help from M. Y. Z. and D. Y. B. M. generated and provided critical material. All authors discussed the results and commented on the manuscript.
The authors declare no competing financial interests. Readers are welcome to comment on the online version of the paper
References
- 1.Martinon F, Burns K, Tschopp J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Molecular Cell. 2002;10:417–426. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 2.Rathinam VA, Vanaja SK, Fitzgerald KA. Regulation of inflammasome signaling. Nat Immunol. 2012;13:333–342. doi: 10.1038/ni.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21:677–687. doi: 10.1038/nm.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martinon F, Mayor A, Tschopp J. Annual Review of Immunology. Annual Review of Immunology. 2009;27:229–265. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- 5.Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol. 2011;29:707–735. doi: 10.1146/annurev-immunol-031210-101405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet. 2001;29:301–305. doi: 10.1038/ng756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feldmann J, et al. Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. American journal of human genetics. 2002;71:198–203. doi: 10.1086/341357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Munoz-Planillo R, et al. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 2013;38:1142–1153. doi: 10.1016/j.immuni.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petrilli V, et al. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death and Differentiation. 2007;14:1583–1589. doi: 10.1038/sj.cdd.4402195. [DOI] [PubMed] [Google Scholar]
- 10.Fry AM, O’Regan L, Sabir SR, Bayliss R. Cell cycle regulation by the NEK family of protein kinases. Journal of cell science. 2012;125:4423–4433. doi: 10.1242/jcs.111195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Richards MW, et al. An autoinhibitory tyrosine motif in the cell-cycle-regulated Nek7 kinase is released through binding of Nek9. Mol Cell. 2009;36:560–570. doi: 10.1016/j.molcel.2009.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yissachar N, Salem H, Tennenbaum T, Motro B. Nek7 kinase is enriched at the centrosome, and is required for proper spindle assembly and mitotic progression. FEBS Lett. 2006;580:6489–6495. doi: 10.1016/j.febslet.2006.10.069. [DOI] [PubMed] [Google Scholar]
- 13.O’Regan L, Fry AM. The Nek6 and Nek7 protein kinases are required for robust mitotic spindle formation and cytokinesis. Mol Cell Biol. 2009;29:3975–3990. doi: 10.1128/MCB.01867-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Salem H, et al. Nek7 kinase targeting leads to early mortality, cytokinesis disturbance and polyploidy. Oncogene. 2010;29:4046–4057. doi: 10.1038/onc.2010.162. [DOI] [PubMed] [Google Scholar]
- 15.Kayagaki N, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–121. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- 16.Kayagaki N, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. 2013;341:1246–1249. doi: 10.1126/science.1240248. [DOI] [PubMed] [Google Scholar]
- 17.Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science. 2013;341:1250–1253. doi: 10.1126/science.1240988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neven B, et al. Molecular basis of the spectral expression of CIAS1 mutations associated with phagocytic cell-mediated autoinflammatory disorders CINCA/NOMID, MWS, and FCU. Blood. 2004;103:2809–2815. doi: 10.1182/blood-2003-07-2531. [DOI] [PubMed] [Google Scholar]
- 19.Meng G, Zhang F, Fuss I, Kitani A, Strober W. A mutation in the Nlrp3 gene causing inflammasome hyperactivation potentiates Th17 cell-dominant immune responses. Immunity. 2009;30:860–874. doi: 10.1016/j.immuni.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kofoed EM, Vance RE. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature. 2011;477:592–595. doi: 10.1038/nature10394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Belham C, et al. A mitotic cascade of NIMA family kinases. Nercc1/Nek9 activates the Nek6 and Nek7 kinases. J Biol Chem. 2003;278:34897–34909. doi: 10.1074/jbc.M303663200. [DOI] [PubMed] [Google Scholar]
- 22.Cohen S, Aizer A, Shav-Tal Y, Yanai A, Motro B. Nek7 kinase accelerates microtubule dynamic instability. Biochimica et biophysica acta. 2013;1833:1104–1113. doi: 10.1016/j.bbamcr.2012.12.021. [DOI] [PubMed] [Google Scholar]
- 23.Kim S, Lee K, Rhee K. NEK7 is a centrosomal kinase critical for microtubule nucleation. Biochem Biophys Res Commun. 2007;360:56–62. doi: 10.1016/j.bbrc.2007.05.206. [DOI] [PubMed] [Google Scholar]
- 24.Ito M, et al. Bruton’s tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nature communications. 2015;6:7360. doi: 10.1038/ncomms8360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sutterwala FS, et al. Critical role for NALP3/CIAS1/cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity. 2006;24:317–327. doi: 10.1016/j.immuni.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 26.Mariathasan S, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 27.Zaki MH, et al. The NLRP3 Inflammasome Protects against Loss of Epithelial Integrity and Mortality during Experimental Colitis. Immunity. 2010;32:379–391. doi: 10.1016/j.immuni.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11:136–140. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
- 29.Schmid-Burgk JL, et al. A genome-wide CRISPR screen identifies NEK7 as an essential component of NLRP3 inflammasome activation. J Biol Chem. 2015 doi: 10.1074/jbc.C115.700492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shi H, et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat Immunol. 2015 doi: 10.1038/ni.3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kanneganti TD, et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature. 2006;440:233–236. doi: 10.1038/nature04517. [DOI] [PubMed] [Google Scholar]
- 32.Wang S, et al. Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE. Cell. 1998;92:501–509. doi: 10.1016/s0092-8674(00)80943-5. [DOI] [PubMed] [Google Scholar]
- 33.He Y, Franchi L, Nunez G. TLR agonists stimulate Nlrp3-dependent IL-1beta production independently of the purinergic P2X7 receptor in dendritic cells and in vivo. J Immunol. 2013;190:334–339. doi: 10.4049/jimmunol.1202737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blasi E, et al. Selective immortalization of murine macrophages from fresh bone marrow by a raf/myc recombinant murine retrovirus. Nature. 1985;318:667–670. doi: 10.1038/318667a0. [DOI] [PubMed] [Google Scholar]
- 35.Swamy M, Siegers GM, Minguet S, Wollscheid B, Schamel WW. Blue native polyacrylamide gel electrophoresis (BN-PAGE) for the identification and analysis of multiprotein complexes. Science’s STKE : signal transduction knowledge environment. 2006;2006:14. doi: 10.1126/stke.3452006pl4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.