

Abstract
Bone morphogenetic proteins (BMPs) induce not only bone formation in vivo but also osteoblast differentiation of mesenchymal cells in vitro. Tumor necrosis factor α (TNFα) inhibits both osteoblast differentiation and bone formation induced by BMPs. However, the molecular mechanisms of these inhibitions remain unknown. In this study, we found that TNFα inhibited the alkaline phosphatase activity and markedly reduced BMP2- and Smad-induced reporter activity in MC3T3-E1 cells. TNFα had no effect on the phosphorylation of Smad1, Smad5, and Smad8 or on the nuclear translocation of the Smad1-Smad4 complex. In p65-deficient mouse embryonic fibroblasts, overexpression of p65, a subunit of NF-κB, inhibited BMP2- and Smad-induced reporter activity in a dose-dependent manner. Furthermore, this p65-mediated inhibition of BMP2- and Smad-responsive promoter activity was restored after inhibition of NF-κB by the overexpression of the dominant negative IκBα. Although TNFα failed to affect receptor-dependent formation of the Smad1-Smad4 complex, p65 associated with the complex. Chromatin immunoprecipitation and electrophoresis mobility shift assays revealed that TNFα suppressed the DNA binding of Smad proteins to the target gene. Importantly, the specific NF-κB inhibitor, BAY11-7082, abolished these phenomena. These results suggest that TNFα inhibits BMP signaling by interfering with the DNA binding of Smads through the activation of NF-κB.
Introduction
Bone morphogenetic proteins (BMPs)2 are members of the transforming growth factor β superfamily (TGF-β) that were originally identified by their ability to induce ectopic bone formation when implanted into muscle tissue (1, 2). BMP signaling is transduced by two types of transmembrane serine-threonine kinase receptor, type I and type II (3, 4). After type II receptors phosphorylate type I receptors in a ligand-dependent fashion, activated type I receptors phosphorylate downstream molecules in the cytoplasm. After BMP type I receptors phosphorylate Smad1, Smad5, and Smad8 (Smad1,5,8), the three Smads form heteromeric complexes with Smad4 and other transcription factors. These complexes translocate into the nucleus and activate the transcription of target genes, including Id1, which encodes an inhibitor of myogenesis (5). This unique and specific ability of BMPs should be useful for the development of bone regeneration. However, BMPs cannot generate enough of a clinical response to be used in bone regeneration (6–8). One possible reason might be that inflammatory cytokines inhibit bone formation and osteoblast differentiation induced by BMPs. For example, several lines of evidence have shown that tumor necrosis factor (TNF) α inhibits osteoblast differentiation in multiple models, including fetal calvaria, bone marrow stromal cells, and MC3T3-E1 cells (9–12).
TNFα is a non-glycosylated protein of 17 kDa, composed of 157 amino acids, that acts as a pleiotropic pro-inflammatory cytokine (13, 14). TNFα is produced primarily by activated macrophages but is also produced by a variety of other structural cell types, including fibroblasts, smooth muscle cells, and osteoblasts (13, 14). Biological responses of TNFα are mediated by the specific binding of either a type I or a type II receptor expressed on the surface of one of many cell types. The binding of TNFα to its receptors results in the activation of an inflammatory response that is classically mediated by a wide variety of pro-inflammatory cytokines, including interleukins, interferon-γ, and chemokines (13, 14). In addition, intracellular signal transduction generated by TNFα elicits a wide spectrum of other cellular responses. These responses include the modulation of the differentiation and proliferation of a variety of cell types and the induction of apoptosis via several signaling pathways, such as the meiosis-specific serine/threonine protein kinase (MEK) pathway, the extracellular signal-regulated kinase (ERK) pathway, the c-Jun N-terminal kinase (JNK) pathway, the p38 kinase pathway, and the NF-κB pathway (14).
Signal transduction in the TNFα pathway occurs partly through the activity of the NF-κB family of transcriptional factors (15, 16). To date, five proteins with conserved homology in the Rel domain (p65 (RelA), RelB, cRel, NF-κB1 (p50/p105), and NF-κB2 (p52/p100)) have been identified. The activity of NF-κB that results from cell stimulation is tightly regulated by the shuttling of NF-κB from the cytoplasm to the nucleus. In unstimulated cells, NF-κB is predominantly localized in the cytoplasm as part of a complex with inhibitory IκB proteins, including IκBα, IκBβ, IκBϵ, and IκBγ. In response to a variety of stimuli, such as TNFα or IL-1β, IκBs are phosphorylated (Ser-32 and Ser-36 for IκBα and Ser-19 and Ser-21 for IκBβ) by the activated IκB kinase (IKK) complex. Phosphorylated IκBs are ubiquitinated and then degraded by the 26 S proteasome. The IKK complex consists of two catalytic kinase subunits, IKKα (IKK1) and IKKβ (IKK2), and a regulatory subunit, NEMO (NF-κB essential modulator), also called IKKγ (15, 16). IKKβ is most critical for the classical (canonical) NF-κB pathway that depends on IκB degradation. In this pathway, the p50/p65 heterodimer enters the nucleus and binds to NF-κB-responsive elements to regulate the expression of genes that are involved in the regulation of immune and inflammatory responses, proliferation, tumorigenesis, and survival (15, 16).
In contrast to NF-κB, BMP signaling provides anti-proliferative differentiation signaling in osteoblasts and in other tissues (17). Furthermore, BMP, via both Smads and Smad-independent mechanisms, inhibits the cell cycle and increases apoptosis by regulating pro-apoptotic proteins (18, 19). Together, the BMP/Smad and NF-κB signaling systems seem to exert antagonistic effects. Therefore, in this study, we assessed the molecular mechanisms underlying TNFα/NF-κB-mediated regulation of BMP-dependent Smad signaling and BMP2-induced osteoblast differentiation.
EXPERIMENTAL PROCEDURES
Reagents
Purified recombinant human BMP2 was provided by Wyeth Pharmaceuticals (Madison, NJ). Recombinant human TNFα was purchased from PeproTech Inc. (Rocky Hill, NJ). Anti-Smad1 (sc-7965), anti-Smad4 (sc-7154), anti-IκBα (sc-371), anti-HDAC1 (sc-7872), anti-β-actin (sc-8432), anti-IgG antibodies p65siRNA (sc-29411), and siRNA transfection reagent (sc-2528) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). BAY11-7082 and anti-p65 antibodies and anti-phosphorylated-Smad1,5,8 antibodies, were obtained from Biomol (Plymouth Meeting, PA) and Cell Signaling (Beverly, MA), respectively.
Cell Culture Conditions
Mouse osteoblastic cell line MC3T3-E1 cells were maintained in an α-modified minimum essential medium (α-MEM) with 10% heat-inactivated fetal bovine serum (FBS) with 100 units/ml penicillin and 100 μg/ml streptomycin. p65-deficient mouse embryonic fibroblasts (p65−/− MEFs) were prepared from p65-deficient mice (20) and maintained in Dulbecco's modified Eagle's medium with 5% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin. These cells were cultured under 5% CO2 at 37 °C.
Alkaline Phosphatase (ALP) Activity and Staining
MC3T3-E1 cells were seeded at a density of 1.0 × 104 cells/well in 96-well plates with α-MEM containing 10% FBS the day before treatment. The cells were pretreated with TNFα (10 ng/ml) for 30 min or left untreated and were subsequently treated with BMP2 (100 ng/ml) for 72 h. After the 72-h treatment, the cells were fixed with an acetone/ethanol mixture (50:50, v/v) and then incubated with a substrate solution (0.1 m diethanolamine, 1 mm MgCl2, and 10 mg/ml p-nitrophenyl phosphate). The reaction was terminated by adding 5 m NaOH, and then the absorbance was measured at 405 nm using a microplate reader (Bio-Rad). To determine ALP activity histochemically, cells were stained for enzyme activity as previously described (5).
Transfection Conditions and Luciferase Assay
MC3T3-E1 cells and p65−/− MEFs were transfected using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. Total amounts of transfected plasmids in each group were equalized by the addition of an empty vector. BMP signaling was monitored using the IdWT4F-luciferase reporter plasmid, which expresses a luciferase protein under the control of a BMP-responsive element in the human ID1 gene, as described previously (21). The pNF-κB luciferase plasmid was purchased from Promega (Madison, WI). Luciferase activities were measured using a dual luciferase system (Promega).
Immunoprecipitation and Immunoblotting
Cells were lysed in TnT buffer (20 mm Tris-HCl, pH 7.5, 200 mm NaCl, 1% Triton X-100, 1 mm dithiothreitol) containing protease inhibitors (Roche, Basel, Switzerland). Cytosolic and nuclear fractions were prepared according to methods described by Dignam et al. (21). For co-precipitation experiments, whole cell extracts were incubated for 6 h at 4 °C with anti-Smad1, anti-Smad4, or anti-p65 antibodies coupled to A/G-Sepharose beads. The immune complex was extensively washed with TnT buffer, and then the samples were boiled and analyzed by immunoblotting. Protein content was measured with Pierce reagent following the manufacturer's protocol. Twenty micrograms of protein were subjected to SDS-polyacrylamide gel electrophoresis (PAGE) and then transferred to a polyvinylidene difluoride membrane at 100 V for 1 h at 4 °C. These membranes were incubated with antibodies at dilutions of 1:500 to 1:1,000 in a 5% dry milk solution containing 0.01% azide overnight at 4 °C. Subsequently, the blots were washed in TTBS (10 mm Tris-HCl, 50 mm NaCl, 0.25% Tween 20) and incubated with a horseradish peroxidase-conjugated secondary antibody. The immunoreactive proteins were visualized using ECL (Amersham Biosciences, Piscataway, NJ).
Chromatin Immunoprecipitation (ChIP) Assays
ChIP was performed with a ChIP Assay Kit (Upstate Biotechnology, Waltham, MA), according to the manufacturer's instructions, using antibodies against phosphorylated Smad1,5,8, p65 antibodies or normal IgG. The purified DNA was analyzed by PCR using primers that amplify sequences containing the Id1 promoter, which harbors a BMP2-responsive element (BRE) to which Smad proteins bind (21). The primer pairs for the Id1, Smad6, and IκBα promoters were 5′-TAAGTTGACCCTTGGTCAGC-3′ (forward) and 5′-GACGTCACCCATTCATAAAAC-3′ (reverse), 5′-CATCCCTAGTGTATCCAACAAAGAG-3′ (forward) and 3′-AGCTCAAGACGGTCACGTACTC-3′ (reverse), and 5′-GCTCATCAAAAAGTTCCCTGTGC-3′ (forward) and 5′-TGGCGAGGTCTGACTGTTGTGG-3′ (reverse), respectively.
Electrophoresis Mobility Shift Assay
EMSAs were performed using MC3T3-E1 cells pretreated with, or without, TNFα (10 ng/ml) for 30 min and were subsequently treated with BMP2 (100 ng/ml) for 30 min. Nuclear extracts were prepared from the pretreated cells. The Smad DNA binding activity in the nuclear fractions was measured by EMSA. The double-stranded BRE probe and the κB probe were radiolabeled using T4 polynucleotide kinase and [γ-32P]ATP (New England Nuclear, Boston, MA) using a labeling kit (Takara Shuzo Co.). The sequences of the oligo DNAs we used were as follows: 5′-TCTCCATGGCGACCGCCCGCGCGGCGCCAGCCT-3′ (BRE probe) and 5′-GATCAGAGGGGACTTTCCGAGG-3′ (κB probe). Six micrograms of the nuclear extracts were incubated with the labeled BRE probe. For the supershift experiments, antibodies were added prior to the addition of the probe, and the mixture was incubated for 15 min at room temperature. Antibodies against Smad1 (sc-7154X) and Smad4 (sc-6201X) were obtained from Santa Cruz Biotechnology. The reaction mixture was loaded onto a 5% polyacrylamide gel in 0.5 × TBE (44.5 mm Tris base, 44.5 mm boric acid, and 1 mm EDTA) and resolved by electrophoresis.
RT-PCR
For semi-quantitative RT-PCR, total RNA form MC3T3-E1 cells prepared with TRIzol (Invitrogen) was amplified by Superscript II and Taq polymerase (Invitrogen). Primer sequences were as follows: Id1: 5′-TCCTGCAGCATGTAATCGAC-3′ (forward) and 5′-GAGAGGGTGAGGCTCTGTTG-3′ (reverse), ALP: 5′-ACTGCTGATCATTCCCACGTT-3′ and (forward), 5′-GAACAGGGTGCGTAGGGAGA-3′ (reverse), osterix: 5′-GCGTCCTCTCTGCTTGA-3′ (forward) and 5′-TGTGTTGCCTGGACCTGGTG-3′ (reverse), and osteocalcin : 5′-AAGCAGGAGGGCAATAAGGT-3′ (forward) and 5′-TTTGTAGGCGGTCTTCAAGC-3′ (reverse); GAPDH: 5′-TGAAGGTCGGTGTGAACGGATTTGGC-3′ (forward) and 5′-CATGTAGGCCATGAGGTCCACCAC-3′ (reverse).
Statistical Analysis
Statistical significance was determined using the Student's t test. A p value of less than 0.05 was considered significant.
RESULTS
TNFα Inhibited BMP2-induced Osteoblast Differentiation
We first examined the effect of TNFα on the BMP2-induced ALP activity, a typical marker of osteoblast differentiation, in MC3T3-E1 cells. BMP2 induced a ∼7-fold increase in ALP activity, and there were many more ALP-positive cells compared with cells cultured with the control levels of BMP2. Consistent with previous reports (10, 11, 22), this induction was completely abolished by pretreatment with TNFα (Fig. 1, A and B).
FIGURE 1.

TNFα inhibited BMP2-induced osteoblast differentiation in MC3T3-E1 cells. A, MC3T3-E1 cells were pretreated with or without TNFα (10 ng/ml) for 30 min and were subsequently treated with BMP2 (100 ng/ml) for 72 h. The cells were fixed with an acetone/ethanol mixture (50:50, v/v) and were then incubated with a substrate solution (0.1 m diethanolamine, 1 mm MgCl2, 10 mg/ml p-nitrophenyl phosphate). ALP activity was then determined. B, cells were stained for ALP activity. C, MC3T3-E1 cells were transiently transfected with the Id1-luc reporter plasmid. After 24 h, the cells were pretreated with or without TNFα (10 ng/ml) for 30 min and were subsequently treated with BMP2 (100 ng/ml) for 24 h. Luciferase activity was determined. Data are means + S.E. (n = 3). Similar results were obtained in three independent experiments. *, p < 0.01.
To precisely determine whether TNFα inhibits BMP signaling via Smad-dependent or Smad-independent mechanisms, we examined the inhibitory effect of TNFα on BMP2-induced Id1-luciferase activity. The Id1-luciferase reporter construct contains a consensus Smad binding site that drives the expression of the luciferase gene (21). BMP2 induced a ∼3–4-fold increase in Id1-luciferase activity compared with controls, and pretreatment with TNFα strongly inhibited the luciferase activity induced by BMP2 (Fig. 1C). These results demonstrate that TNFα inhibits osteoblast differentiation and BMP2-induced Id1-luciferase activity by inhibiting the Smad-dependent pathway.
TNFα Did Not Affect the Phosphorylation or Nuclear Translocation of Smad1,5,8 Induced by BMP2
Upon binding of the BMP ligand to the type I and type II receptor complexes, the activated type I receptor phosphorylates Smad1,5,8, which then assemble into complexes with Smad4 and translocate into the nucleus. In the nucleus, the Smad complexes regulate the expression of genes related to osteoblast differentiation, such as ALP and osteocalcin (24, 25). The previous report demonstrated that TNFα inhibited BMP-induced Smad1,5,8 phosphorylation in C2C12 cells (23). In this study, as shown in Fig. 2A, phosphorylation of Smad1,5,8 was first detected within 10 min and declined thereafter (Fig. 2A). Pretreatment with TNFα did not affect the Smad1,5,8 phosphorylation induced by BMP2 (Fig. 2A). Furthermore, the translocation of phosphorylated Smad1,5,8 was not inhibited by pretreatment with TNFα, although TNFα led to nuclear translocation of p65, an NF-κB subunit (Fig. 2B).
FIGURE 2.

TNFα did not affect the phosphorylation or the nuclear translocation of Smad1,5,8 induced by BMP2. A, MC3T3-E1 cells were pretreated with or without TNFα (10 ng/ml) for 30 min and were subsequently treated with BMP2 (100 ng/ml) for the indicated time. Total cell lysates were immunoblotted with anti-phosphorylated Smad1,5,8 antibodies, and anti-β-actin was used as a loading control. B, MC3T3-E1 cells were pretreated with or without TNFα (10 ng/ml) for 30 min and were subsequently treated with BMP2 (100 ng/ml) for 1 h. Cytosolic and nuclear fractions were prepared, and then lysates were immunoblotted with anti-phosphorylated Smad1,5,8, and anti-p65 antibodies. Anti-IκBα antibodies and anti-HDAC antibodies were used to probe the cytosolic fraction and nuclear fraction, respectively. Similar results were obtained in three independent experiments.
Activation of NF-κB Inhibited BMP2-induced Id1-luciferase Activity in Both MC3T3-E1 Cells and p65−/− MEFs
We hypothesized that TNFα inhibits BMP/Smad signaling through NF-κB activation because NF-κB regulates the expression of several genes related to inflammation induced by TNFα (15, 16). The most abundant form of NF-κB is a heterodimer of the p50 and p65 subunits that contains a transcriptional activator domain (15, 16). p65−/− MEFs transfected with Id1-luc or pNF-κB-luc were cotransfected together with or without p65 and were treated with BMP2 for 24 h. The transcriptional activity of NF-κB was evaluated in a p65 dose-dependent manner (Fig. 3A). The inhibition of BMP2-induced Id1-luciferase activity coincided with the activation of NF-κB in p65−/− MEFs (Fig. 3B). To further confirm the possibility that NF-κB activation inhibits BMP-induced Id1-luc activity, we inhibited NF-κB activity in MC3T3-E1 cells by two different approaches: using p65 siRNA to knockdown p65 expression or using the pharmacological NF-κB-specific inhibitor BAY11-7082. BMP2-induced Id1-luc activity was reduced by pretreatment with TNFα in MC3T3-E1 cells (Fig. 3C, lanes 3 and 4). By contrast, TNFα failed to suppress BMP2-induced Id1-luc activity in MC3T3-E1 cells that had been transfected with p65 siRNA (Fig. 3C, lanes 4 and 8). NF-κB suppression by BAY11-7082 also led to a recovery of Id1-luc activity (Fig. 3C, lanes 4 and 12).
FIGURE 3.

Activation of NF-κB inhibited BMP2-induced Id1-luciferase activity in MC3T3-E1 cells and p65−/− MEFs. A, p65−/− MEFs were transiently transfected with a pNF-κB-luc reporter and increasing concentrations of a p65 expression plasmid and were then assayed for luciferase activity after 24 h. B, p65−/− MEFs were transiently transfected with an Id1-luc reporter with or without a p65 expression plasmid for 24 h. The cells were then treated with BMP2 (100 ng/ml) and assayed for luciferase activity after 24 h. C, NF-κB activation in MC3T3-E1 cells was inhibited by either p65 siRNA or BAY11-7082, and then the cells were treated with BMP2 (100 ng/ml) with or without pretreatment with TNFα (10 ng/ml). The cells were assayed for Id1- or NF-κB- luciferase activity after 24 h. The expression level of p65 was examined by immunoblotting using anti-p65 antibodies.
Furthermore, we cotransfected cells with expression constructs for Id1-luc, a constitutively active form of FLAG-Smad1 (FLAG-Smad1(DVD)) that exchanges two distal serines that are part of the C-terminal SSXS motif for aspartic acids (4), FLAG-Smad4, and/or p65, with or without an expression vector for a dominant-negative form of IκBα (IκBαDN). The overexpression of p65 completely abrogated the Id1-luciferase activity induced by FLAG-Smad1(DVD) and FLAG-Smad4. By contrast, the transient overexpression of IκBαDN, which inhibits NF-κB in the cytoplasm, rescued this inhibition (Fig. 4A). TNFα signaling can activate MAPK pathway members, including MEK, ERK, and JNK (14). Recently, MAPK phosphorylation sites were identified in Smad1 at Ser-187, Ser-195, Ser-206, and Ser-214. Subsequently, the phosphorylation of these sites by MAPK negatively regulates BMP signaling (26). Therefore, we mutated all of these serines to alanines to get the FLAG-Smad1(MAPKmut-DVD) construct. The overexpression of p65 strongly inhibited the Id1-luciferase activity induced by FLAG-Smad1(MAPKmut-DVD) and FLAG-Smad4. Furthermore, the transient overexpression of IκBαDN again rescued this inhibition (Fig. 4B). These results suggest that cross-talk between NF-κB and BMP/Smad signaling influences BMP2-induced osteoblast differentiation induced by pretreatment with TNFα.
FIGURE 4.
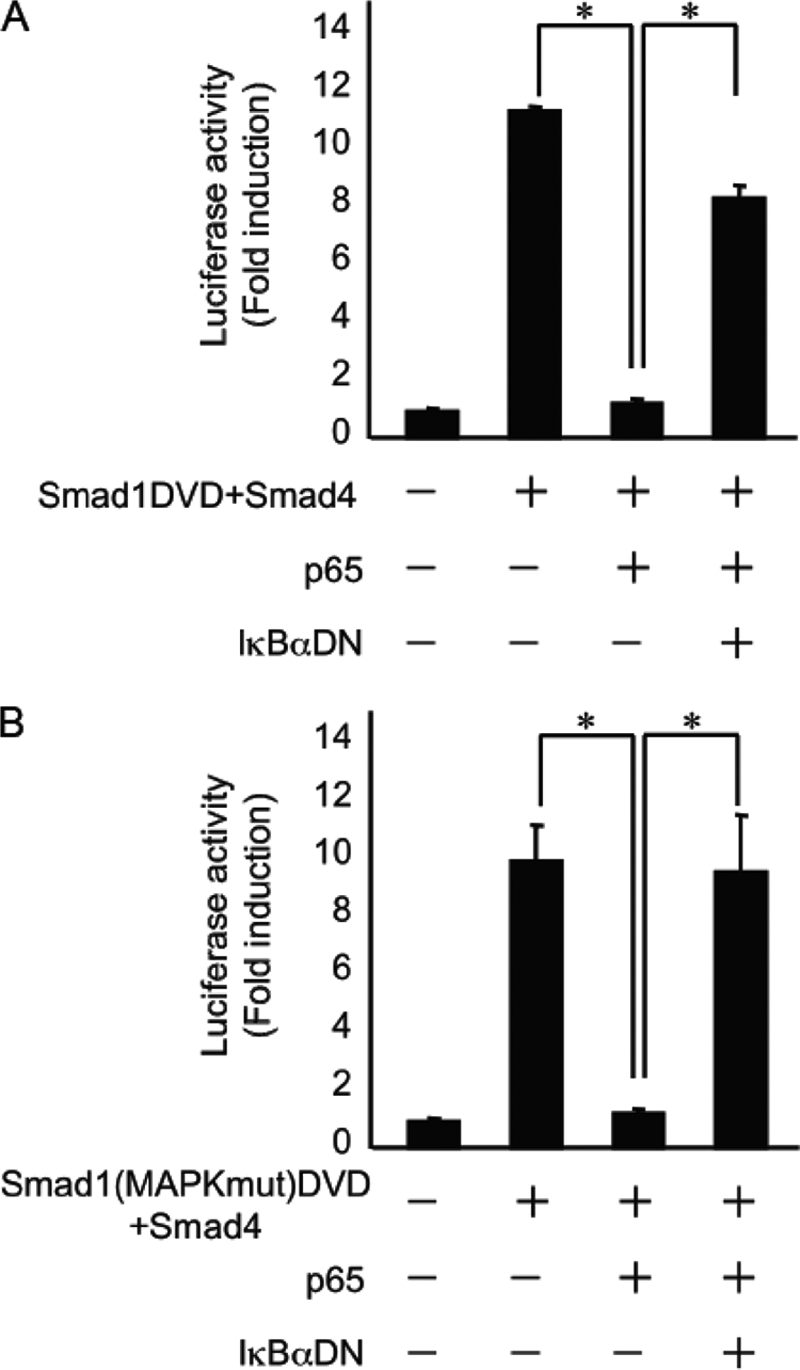
Overexpression of p65 inhibited Id1-luciferase activity induced by the constitutively active form of Smad1-induced in p65−/− MEFs. p65−/− MEFs were transiently transfected with Id1-luc, FLAG-Smad1(DVD) (A) or with FLAF-Smad1(MAPKmut-DVD) (B), FLAG-Smad4, p65, and IκBαDN expression plasmids and were assayed for luciferase activity after 24 h. Data are means + S.E. (n = 3). Similar results were obtained in three independent experiments. *, p < 0.01.
TNFα Did Not Disrupt the Smad1 and Smad4 Complex but Did Activate p65 through the Association of TNFα with Smad1 and Smad4
To confirm the possibility that TNFα interferes with the formation of a Smad1/Smad4 complex, we examined the interactions between Smad1 and Smad4 in cells pretreated with or without TNFα using co-immunoprecipitation assays. Cell extracts were subjected to immunoprecipitation with anti-Smad1, anti-Smad4, or anti-p65 antibodies, followed by immunoblot analysis with individual antibodies. Smad1 and Smad4 formed a complex in a ligand-dependent fashion (Fig. 5A, lanes 2 and 7). Pretreatment with TNFα failed to disrupt the association of Smad1 and Smad4. However, p65 activated by TNFα did associate with the complex of Smad1 and Smad4 (Fig. 5A, lanes 3, 8, and 13). Pretreatment with BAY11-7082 interfered with the association of p65 with Smad1 and/or Smad4 but did not interfere with the complex of Smad1 and Smad4 without affecting the expression levels of Smad1, Smad4, and p65 (Fig. 5, A, lanes 4, 5, 9, 10, 14, and 15 and B).
FIGURE 5.

TNFα did not disrupt the Smad1 and Smad4 complex but activated p65 by TNFα associated with Smad1 and Smad4. A, MC3T3-E1 cells were pretreated with or without TNFα (10 ng/ml) for 30 min and were subsequently treated with BMP2 (100 ng/ml) for 1 h in the presence or absence of increasing amounts of BAY11-7082. The whole cell extract was immunoprecipitated with anti-Smad1, anti-Smad4, or anti-p65 antibodies that were coupled to A/G-Sepharose beads; the whole cell extract was then processed for immunoblotting with the indicated antibodies. B, part of the whole cell extract was immunoblotted with the indicated antibodies to examine that all treatments did not affect the expression levels of Smad1, Smad4, and p65 and anti-β-actin was used as a loading control. Similar results were obtained in three independent experiments.
Activation of p65 by TNFα Inhibits the DNA Binding Activities of Smad Proteins Induced by BMP2
We next examined whether TNFα inhibited the DNA binding of Smad complexes induced by BMP2. MC3T3-E1 cells pretreated with or without TNFα were treated with BMP2 for the indicated time. Following pretreatment, genomic DNA was immunoprecipitated with anti-phosphorylated Smad1,5,8 antibodies, or control IgG, and was subsequently subjected to PCR amplification using primers that amplify the Id1 promoter region harboring the BMP2-responsive element. ChIP analysis showed that phosphorylated Smad1,5,8 was recruited to the Id1 promoter after 30 min of BMP2 stimulation, reached a maximum level at 1 h, and declined thereafter (Fig. 6A). When cells were pretreated with TNFα, the recruitment of phosphorylated Smad1,5,8 to the Id1 promoter was strongly suppressed compared with BMP2 treatment alone (Fig. 6A). p65 still bound to the Id1 promoter under these conditions (Fig. 6A). Similar results were obtained using Smad6, a BMP target gene (27), promoter (Fig. 6B).
FIGURE 6.

Activation of p65 by TNFα inhibited the BMP-induced Smad1 recruitment to the Id1 and Smad6 promoters. MC3T3-E1 cells were pretreated with or without TNFα (10 ng/ml) for 30 min and were subsequently treated with BMP2 (100 ng/ml) for the indicated time. Chromatin from individual samples was precipitated using the indicated antibodies or with control IgG. The Id1 promoter (A) or the Smad6 promoter (B) was amplified by PCR from the precipitated DNA.
To further confirm whether the inhibitory effect of TNFα on the activation of Smad-DNA binding in response to BMP2 is mediated by p65, we performed EMSAs using nuclear extracts prepared from MC3T3-E1 cells. BMP2 induced the formation of DNA-binding protein complexes in MC3T3-E1 cells (Fig. 7A, lane 3). Consistent with the ChIP analysis, TNFα suppressed BMP2-induced DNA-binding protein complexes (Fig. 7A, lanes 3 and 4). The addition of anti-Smad1 or anti-Smad4 antibodies to the BMP2-treated nuclear extracts reduced the amount of the complex and induced supershifted bands (Fig. 7B, lanes 3 and 4). Moreover, when both the anti-Smad1 and anti-Smad4 antibodies were added together, a slower-migrating supershifted band appeared (Fig. 7B). We further performed EMSAs using nuclear extracts prepared from p65−/− MEFs. BMP2 induced Smad DNA binding in wild-type and p65−/− MEFs (Fig. 7C, lanes 2 and 5). TNFα pretreatment in wild-type strongly suppressed the DNA binding activities of Smad proteins induced by BMP2 (Fig. 7C, lanes 3 and 6). Although TNFα slightly suppressed Smad DNA binding activity, large amount of Smads still bound to DNA in p65−/− MEFs compared with wild-type cells (Fig. 7C, lanes 5 and 6). TNFα induced NF-κB DNA binding in wild-type but not p65−/− MEFs (Fig. 7C, lanes 9 and 12). These results suggest that the activation of p65 by TNFα inhibits the DNA binding of Smad proteins normally induced by BMP2.
FIGURE 7.

Activation of p65 by TNFα inhibited the DNA-binding activities of Smad proteins induced by BMP2. A, MC3T3-E1 cells were pretreated with or without increasing amounts of BAY11-7082 and TNFα (10 ng/ml) for 30 min and were subsequently treated with BMP2 (100 ng/ml) for 30 min. The BMP2-induced DNA binding activity in the nuclear fractions was measured by EMSA using a BRE probe. B, subunit composition of the BMP2-induced DNA-binding proteins in MC3T3-E1 cells. The nuclear extracts were pretreated at 4 °C for 60 min with vehicle (lanes 1 and 2) or with 1 μl of the following polyclonal antibodies: anti-Smad1 (lane 3), anti-Smad4 (lane 4), anti-Smad1 and anti-Smad4 (lane 5), and control IgG (lane 6) antibodies. EMSAs were performed on these pretreated nuclear extracts. C, wild-type or p65−/− MEFs were pretreated with or without TNFα (10 ng/ml) for 30 min and were subsequently treated with BMP2 (100 ng/ml) for 30 min. The Smad and NF-κB DNA binding activities in the nuclear fractions were measured by EMSA using a BRE probe and a κB probe, respectively. Similar results were obtained in three independent experiments.
A Specific Inhibitor of NF-κB Signaling Restored the TNFα-induced Inhibition of the DNA Binding Activities of Smad Proteins and the ALP Activity Induced by BMP2
Because NF-κB signaling was found to antagonize Smad activation by BMP2, we next examined the effect of BAY11-7082 on the TNFα-induced inhibition of the DNA binding activities of Smad proteins by BMP2. NF-κB suppression by BAY11-7082 led to a recovery of the binding of phosphorylated Smad1,5,8 to the Id1 promoter (Fig. 8A). By contrast, TNFα recruited p65 to the IκBα promoter, a target gene of NF-κB, but BAY11-7082 suppressed this recruitment in a dose-dependent manner (Fig. 8A). BAY11-7082 also rescued the inhibitory effect of TNFα on BMP2-induced Smad DNA binding in a dose-dependent manner (Fig. 7A, lanes 4–6). We next examined the expression of osteoblast differentiation marker genes in MC3T3-E1 cells. BMP2 induced Id1, ALP, osterix, and osteocalcin mRNA expression within 12 h, whereas TNFα pretreatment strongly suppressed the expression of these genes. BAY11-7082 restored TNFα-mediated inhibition of gene expression in a dose-dependent manner (Fig. 8B). In addition, the TNFα-mediated inhibition of ALP activity and the staining induced by BMP2 were also restored by pretreatment with BAY11-7082 (Fig. 8, C and D). These results strongly suggest that TNFα represses BMP signaling by interfering with the DNA binding of Smads through the activation of NF-κB.
FIGURE 8.

A specific inhibitor of NF-κB signaling restored the TNFα-induced inhibition of the DNA binding activities of Smad proteins, expression of osteoblast marker genes, and ALP activity, which were all induced by BMP2. A, MC3T3-E1 cells were pretreated with BAY11-7082 and TNFα (10 ng/ml) for 30 min and were subsequently treated with BMP2 (100 ng/ml) for the indicated time. Chromatin from individual samples was precipitated using the indicated antibodies or control IgG. The Id1 or IκBα promoter was amplified by PCR from the precipitated DNA. B, MC3T3-E1 cells were pretreated with BAY11-7082 and TNFα (10 ng/ml) for 30 min and were subsequently treated with BMP2 (100 ng/ml) for 12 h. Total RNA was extracted and subjected to reverse transcription with a random primer. The first strand cDNAs were submitted to PCR analysis for Id-1, ALP, osterix, osteocalcin, and GAPDH. C, MC3T3-E1 cells were pretreated with BAY11-7082 and TNFα (10 ng/ml) for 30 min and were subsequently treated with BMP2 (100 ng/ml) for 72 h. The cells were fixed with an acetone/ethanol mixture (50:50, v/v) and were incubated with a substrate solution (0.1 m diethanolamine, 1 mm MgCl2, 10 mg/ml p-nitrophenyl phosphate). ALP activity was then determined. Data are means + S.E. (n = 3). Similar results were obtained in three independent experiments. *, p < 0.01. D, The cells were stained for ALP activity.
DISCUSSION
TNFα is a potent inflammatory cytokine that contributes to local and systemic bone loss in inflammatory bone diseases such as rheumatoid arthritis and periodontitis, as well as in estrogen deficiency (28–31). In patients with rheumatoid arthritis, TNFα and other cytokines are overproduced in inflamed joints by various cells that infiltrate the synovial membrane (31). Anti-TNF drugs such as infliximab, etanercept, and adalimumab have been shown not only to diminish signs and symptoms of disease, but also to prevent joint damage (32). Furthermore, elevated production of TNFα in postmenopausal women and in animal models of postmenopausal osteoporosis augments bone destruction by stimulating osteoclastic bone resorption (29, 33). Under these conditions, osteoblast-mediated bone formation cannot compensate for accelerated osteoclastic bone resorption. These findings suggest a direct inhibitory effect of TNFα on osteoblasts. Further support for this inhibitory effect comes from research showing that although TNFα transgenic mice, a well-established animal model of rheumatoid arthritis, exhibit well-described features of rheumatoid arthritis, these animals also develop symptoms of general osteoporosis, such as a reduction in trabecular bone in the metaphysic, as compared with wild-type littermates (34). In addition, osteoblastic cells derived from TNFα transgenic mice form significantly fewer and smaller nodules under basal conditions and in the presence of BMP2, again indicating that TNFα reduces osteoblast function. Not only in pathological conditions, but also at the endogenous level, the level of TNFα present is of a magnitude sufficient to reduce bone formation, leading to a decrease in the maximum achievable peak bone mineral density (BMD) and bone mass. Both TNFα- and TNF type I receptor-deficient mice exhibit higher BMDs than wild-type littermates (35).
The inhibitory effects of TNF on bone formation in vitro were first reported in 1987 using a neonatal rat calvarial organ culture system (9). Subsequent studies demonstrated that TNFα blocks osteoblast differentiation in multiple models including fetal calvaria, bone marrow stromal cells, and MC3T3-E1 cells (10–12). TNFα treatment of fetal calvaria precursor cells, which spontaneously acquire the osteoblast phenotype over 21 days, inhibited differentiation, as shown by reduced formation of mineralizing nodules and decreased secretion of osteocalcin, a mature osteoblast marker. TNFα inhibited the expression of insulin-like growth factor I (IGF-I) but not the expression of BMP2, BMP4 or BMP6. Also, the addition of IGF-I or BMP6 could not rescue this TNF inhibition, suggesting that TNF acted downstream of these proteins in the differentiation pathway (11).
TNFα inhibited not only spontaneous osteoblast differentiation, but also BMP-induced osteoblast differentiation via Smad or via Smad-independent mechanisms (23). TNFα inhibited the BMP2-induced expression of Runx and osteocalcin and BMP2-induced ALP activity in C2C12 cells, a mouse myoblastic cell line, in a dose-dependent manner. TNFα inhibited phosphorylation of Smad1,5,8 by inducing Smad6 expression, an inhibitory Smad (23). We did not observe an inhibitory effect of TNFα on phosphorylation nor on translocation into the nucleus of Smad1,5,8 induced by BMP2 in MC3T3-E1. We treated cells with TNFα for 30 min and did not observe any induction of Smad6 or Smad7 by the TNFα treatment (data not shown). Although we could not explain this discrepancy, the discrepancy might depend on the stage of osteoblast differentiation and in the type of cell. Pretreatment with TNFα did inhibit the Smad-dependent Id1-luciferase activity induced by BMP2. These results strongly suggest that TNFα inhibits BMP/Smad signaling in the nucleus by interfering with the DNA binding or transcriptional modification of Smad proteins.
TNF signals through multiple intracellular pathways. In osteoblasts, a TNF trimer binds two receptor forms, type I and type II, of which only type I mediates the inhibition of osteoblast differentiation (36). In a well-established paradigm, the bound receptor activates MAP kinase or NF-κB signals (13, 14). We focused on NF-κB signals, because BMP and NF-κB have opposite biological functions during inflammatory processes (15, 16, 18, 19). We clearly showed that NF-κB is involved in the inhibitory effect of TNFα on osteoblast differentiation that is induced by BMP2. First, the inhibition of BMP2-induced Id1-luciferase activity coincided with the activation of NF-κB in p65−/− MEFs in a p65 dose-dependent manner. The overexpression of p65 completely abrogated the Id1-luciferase activity induced by the constitutively active forms of Smad1 and Smad4. Second, although TNFα inhibited BMP-induced DNA binding of Smad proteins in wild-type MEFs, TNFα failed to inhibit the DNA binding in p65−/− MEFs. Finally, a pharmacological NF-κB inhibitor restored the inhibitory effects of TNFα on both the BMP-induced DNA binding of Smad proteins and ALP activity. Whereas others have shown that NF-κB antagonizes BMP/Smad signaling by enhancing Smad7 expression (37), this study suggests that NF-κB, particularly the p65 subunit, binds the Smad1 and Smad4 complex, directly or indirectly, and that this binding interferes with the DNA binding of Smad proteins induced by BMP2.
There are some reports that TNFα inhibits BMP signaling by signals other than NF-κB. TNFα promotes Runx2 degradation through the up-regulation of Smurf1 and Smurf2 in osteoblasts, leading to suppressed osteoblast differentiation (34). Inhibitors of MEK and ERK, but not inhibitors of JNK or p38 kinase, abrogate TNFα inhibition of osterix mRNA and promoter activity (12). Moreover, TNFα acts to inhibit BMP signaling, including Smad1,5,8 phosphorylation and Id1 transcription, through activating the JNK pathway (23). These results suggest that TNFα inhibits osteoblast differentiation by inhibiting multiple steps that are required for differentiation from osteoblast progenitors to mature osteoblasts by interfering with several signals downstream of the TNF type I receptor.
Two transcriptional factors, Runx2 (Cbfa1/AML3/Pebp2αA) and osterix (Osx/Sp7), are required for differentiation of the osteoblast lineage (38, 39). The in vivo significance of these two factors has been verified by the observations that targeting disruption of either gene in mice results in a complete lack of both endochondral and intramembranous ossification and that these mice are characterized by an absence of mature osteoblasts throughout the body (38, 39). TNFα inhibits bone formation by promoting Runx2 proteasomal degradation through the up-regulation of Smurf1 and Smurf2 expression (34, 40). Furthermore, TNF inhibits the expression of osterix mRNA by suppressing osterix promoter activity via the MAP kinase and NF-κB pathways (41). These results indicate that TNFα inhibits osteoblast differentiation by suppressing the master regulators of osteoblast differentiation, such as Runx2 and osterix.
Collectively, our present study reveals that the inhibitory effect of TNFα on BMP signaling in the process of osteoblast differentiation is closely linked to the NF-κB pathway. Smad1,5,8 signaling is now generally accepted as a very early downstream effector in the BMP signaling pathway and is thus fundamental for osteoblastic differentiation in response to BMP ligands. The impairment of this central activation step, which is evoked by TNFα, is critical for bone differentiation and cellular sensitivity to BMP ligands. TNFα ablates osteoblast differentiation, at least in part, through NF-κB activation and subsequent interference with the DNA binding of Smad proteins. An intracellular balance of the signal intensities between TNFα/NF-κB and BMP/Smad is therefore crucial for osteoblast differentiation and could also be useful for bone regeneration.
Acknowledgment
We thank Kazuhiro Kanomata for technical assistance.
This work was supported in part by a grant-in-aid from Kyushu Dental College Internal Grants (to H. F. and E. J.), a grant-in-aid from the Ministry of Education, Culture, Sports, Science and Technology of Japan (to E. J.: 20390473), and by the Astellas Foundation for Research on Metabolic Disorders (to E. J.).
- BMP
- bone morphogenetic protein
- TNFα
- tumor necrosis factor α
- NF-κB
- nuclear factor κB
- ALP
- alkaline phosphatase
- EMSA
- electrophoretic mobility shift assay
- BRE
- BMP2-responsive element
- MEF
- mouse embryonic fibroblast cells.
REFERENCES
- 1.Urist M. R. (1965) Science 150, 893–899 [DOI] [PubMed] [Google Scholar]
- 2.Wozney J. M., Rosen V., Celeste A. J., Mitsock L. M., Whitters M. J., Kriz R. W., Hewick R. M., Wang E. A. (1988) Science 242, 1528–1534 [DOI] [PubMed] [Google Scholar]
- 3.Miyazono K., Maeda S., Imamura T. (2005) Cytokine Growth Factor Rev. 16, 251–263 [DOI] [PubMed] [Google Scholar]
- 4.Massagué J., Seoane J., Wotton D. (2005) Genes Dev. 19, 2783–2810 [DOI] [PubMed] [Google Scholar]
- 5.Katagiri T., Yamaguchi A., Komaki M., Abe E., Takahashi N., Ikeda T., Rosen V., Wozney J. M., Fujisawa-Sehara A., Suda T. (1994) J. Cell Biol. 127, 1755–1766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kusumoto K., Bessho K., Fujimura K., Akioka J., Okubo Y., Wang Y., Iizuka T., Ogawa Y. (2002) J. Int. Med. Res. 30, 251–259 [DOI] [PubMed] [Google Scholar]
- 7.Vaidya R., Sethi A., Bartol S., Jacobson M., Coe C., Craig J. G. (2008) J. Spinal Disord. Tech. 21, 557–562 [DOI] [PubMed] [Google Scholar]
- 8.Buma P., Arts J. J., Gardeniers J. W., Verdonschot N., Schreurs B. W. (2008) J. Biomed. Mater. Res. B Appl. Biomater. 84, 231–239 [DOI] [PubMed] [Google Scholar]
- 9.Canalis E. (1987) Endocrinology 121, 1596–1604 [DOI] [PubMed] [Google Scholar]
- 10.Nakase T., Takaoka K., Masuhara K., Shimizu K., Yoshikawa H., Ochi T. (1997) Bone 21, 17–21 [DOI] [PubMed] [Google Scholar]
- 11.Gilbert L., He X., Farmer P., Boden S., Kozlowski M., Rubin J., Nanes M. S. (2000) Endocrinology 141, 3956–3964 [DOI] [PubMed] [Google Scholar]
- 12.Nanes M. S. (2003) Gene 321, 1–15 [DOI] [PubMed] [Google Scholar]
- 13.Chen G., Goeddel D. V. (2002) Science 296, 1634–1635 [DOI] [PubMed] [Google Scholar]
- 14.Bradley J. R. (2008) J. Pathol. 214, 149–160 [DOI] [PubMed] [Google Scholar]
- 15.Ghosh S., Hayden M. S. (2008) Nat. Rev. Immunol. 8, 837–848 [DOI] [PubMed] [Google Scholar]
- 16.Karin M. (2008) Cell Res. 18, 334–342 [DOI] [PubMed] [Google Scholar]
- 17.ten Dijke P. (2006) Curr. Med. Res. Opin. 22, S7–11 [DOI] [PubMed] [Google Scholar]
- 18.Haÿ E., Lemonnier J., Fromigué O., Marie P. J. (2001) J. Biol. Chem. 276, 29028–29036 [DOI] [PubMed] [Google Scholar]
- 19.Haÿ E., Lemonnier J., Fromigué O., Guénou H., Marie P. J. (2004) J. Biol. Chem. 279, 1650–1658 [DOI] [PubMed] [Google Scholar]
- 20.Doi T. S., Takahashi T., Taguchi O., Azuma T., Obata Y. (1997) J. Exp. Med. 185, 953–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Katagiri T., Imada M., Yanai T., Suda T., Takahashi N., Kamijo R. (2002) Genes Cells 7, 949–960 [DOI] [PubMed] [Google Scholar]
- 22.Dignam J. D., Lebovitz R. M., Roeder R. G. (1983) Nucleic Acids Res. 11, 1475–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mukai T., Otsuka F., Otani H., Yamashita M., Takasugi K., Inagaki K., Yamamura M., Makino H. (2007) Biochem. Biophys. Res. Commun. 356, 1004–1010 [DOI] [PubMed] [Google Scholar]
- 24.Yamamoto N., Akiyama S., Katagiri T., Namiki M., Kurokawa T., Suda T. (1997) Biochem. Biophys. Res. Commun. 238, 574–580 [DOI] [PubMed] [Google Scholar]
- 25.Nishimura R., Hata K., Harris S. E., Ikeda F., Yoneda T. (2002) Bone 31, 303–312 [DOI] [PubMed] [Google Scholar]
- 26.Sapkota G., Alarcón C., Spagnoli F. M., Brivanlou A. H., Massagué J. (2007) Mol. Cell 25, 441–454 [DOI] [PubMed] [Google Scholar]
- 27.Ishida W., Hamamoto T., Kusanagi K., Yagi K., Kawabata M., Takehara K., Sampath T. K., Kato M., Miyazono K. (2000) J. Biol. Chem. 275, 6075–6079 [DOI] [PubMed] [Google Scholar]
- 28.Ralston S. H., Russell R. G., Gowen M. (1990) J. Bone Miner. Res. 5, 983–988 [DOI] [PubMed] [Google Scholar]
- 29.Pacifici R., Brown C., Puscheck E., Friedrich E., Slatopolsky E., Maggio D., McCracken R., Avioli L. V. (1991) Proc. Natl. Acad. Sci. U.S.A. 88, 5134–5138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ammann P., Rizzoli R., Bonjour J. P., Bourrin S., Meyer J. M., Vassalli P., Garcia I. (1997) J. Clin. Invest. 99, 1699–1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choy E. H., Panayi G. S. (2001) N. Engl. J. Med. 344, 907–916 [DOI] [PubMed] [Google Scholar]
- 32.Ranganathan P. (2008) Curr. Opin. Mol. Ther. 10, 562–567 [PubMed] [Google Scholar]
- 33.Cenci S., Weitzmann M. N., Roggia C., Namba N., Novack D., Woodring J., Pacifici R. (2000) J. Clin. Invest. 106, 1229–1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaneki H., Guo R., Chen D., Yao Z., Schwarz E. M., Zhang Y. E., Boyce B. F., Xing L. (2006) J. Biol. Chem. 281, 4326–4333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Y., Li A., Strait K., Zhang H., Nanes M. S., Weitzmann M. N. (2007) J. Bone Miner. Res. 22, 646–655 [DOI] [PubMed] [Google Scholar]
- 36.Gilbert L. C., Rubin J., Nanes M. S. (2005) Am. J. Physiol. Endocrinol. Metab. 288, E1011–E1018 [DOI] [PubMed] [Google Scholar]
- 37.Eliseev R. A., Schwarz E. M., Zuscik M. J., O'Keefe R. J., Drissi H., Rosier R. N. (2006) Exp. Cell Res. 312, 40–50 [DOI] [PubMed] [Google Scholar]
- 38.Komori T., Yagi H., Nomura S., Yamaguchi A., Sasaki K., Deguchi K., Shimizu Y., Bronson R. T., Gao Y. H., Inada M., Sato M., Okamoto R., Kitamura Y., Yoshiki S., Kishimoto T. (1997) Cell 89, 755–764 [DOI] [PubMed] [Google Scholar]
- 39.Nakashima K., Zhou X., Kunkel G., Zhang Z., Deng J. M., Behringer R. R., de Crombrugghe B. (2002) Cell 108, 17–29 [DOI] [PubMed] [Google Scholar]
- 40.Gilbert L., He X., Farmer P., Rubin J., Drissi H., van Wijnen A. J., Lian J. B., Stein G. S., Nanes M. S. (2002) J. Biol. Chem. 277, 2695–2701 [DOI] [PubMed] [Google Scholar]
- 41.Lu X., Gilbert L., He X., Rubin J., Nanes M. S. (2006) J. Biol. Chem. 281, 6297–6306 [DOI] [PubMed] [Google Scholar]