

Summary
Activation of NF-kB has been noted in many tumor types, however only rarely has this been linked to an underlying genetic mutation. An integrated analysis of high-density oligonucleotide array CGH and gene expression profiling data from 155 multiple myeloma samples identified a promiscuous array of abnormalities contributing to the dysregulation of NF-kB in approximately 20% of patients. We report mutations in ten genes causing the inactivation of TRAF2, TRAF3, CYLD, cIAP1/cIAP2, and activation of NFKB1, NFKB2, CD40, LTBR, TACI, and NIK that result primarily in constitutive activation of the non-canonical NF-kB pathway, with the single most common abnormality being inactivation of TRAF3. These results highlight the critical importance of the NF-kB pathway in the pathogenesis of multiple myeloma.
Significance
Activation of the canonical NF-kB pathway has been recognized in patients with multiple myeloma, and attributed to interactions of the myeloma cell with the bone marrow microenvironment. We report a promiscuous array of mutations that result in constitutive activation of primarily the non-canonical NF-kB pathway in one fifth of patients. The genetic selection of these mutations by the tumor cells highlights the critical importance of the NF-kB pathway in myeloma and suggests an important mechanism by which the tumor cells can begin to become independent of the microenvironment. Our results suggest that proteasome inhibitors, that target the NF-kB pathway, may be most effective in those patients with mutations causing constitutive activation of NF-kB.
Introduction
Multiple myeloma (MM) is a late stage B-cell malignancy characterized by the accumulation of a monoclonal plasma cell population in the bone marrow. One half of all patients have recurrent immunoglobulin gene translocations, mediated by aberrant class switch recombination events, while the other half are hyperdiploid (Bergsagel et al., 1996; Fonseca et al., 2003b). However, both groups are characterized by dysregulation of a cyclin D gene, which appears to be an early and unifying event in MM pathogenesis (Bergsagel et al., 2005). A variety of secondary genetic events, associated with disease progression have been observed and include deletion of chromosome 13, amplification of chromosome 1q, deletion of TP53, activating point mutations of ras, and translocations of c-myc (Kuehl and Bergsagel, 2002). In addition to these known genetic events, the activation of NF-kB, by mechanisms that remain unclear, is thought to be a common event in MM. The activation of the NF-kB pathway by mutation has only been described in a single MM patient with an activating mutation of NFKB2 (Fracchiolla et al., 1993). Nevertheless, the importance of this pathway in MM is highlighted by the clinical success of proteasome inhibitors (Richardson et al., 2003), which have been postulated to act principally by inhibiting NF-kB (Rajkumar et al., 2005).
The transcriptional regulation of NF-kB target genes is mediated by an array of homo- and hetero-dimers containing p50 and p52, the active isoforms of NFKB1(p105) and NFKB2(p100) respectively (Hoffmann and Baltimore, 2006). The function of these dimers is dependent on ubiquitin mediated proteasomal degradation; however, the regulation of this process is different for dimers containing p50 or p52. The classical or “canonical” NF-kB pathway is negatively regulated by the inhibitors of NF-kB (IkBs), which retain p50-containing complexes in the cytoplasm to prevent their function as transcription factors. Therefore, the “canonical” pathway is not regulated by the processing of NFKB1 as the inactivation of the IkB complex is the rate-limiting step in the activation of the “canonical” pathway. In contrast, the “non-canonical” NF-kB pathway is solely regulated by the control of NFKB2(p100) processing to the active p52 isoform (Dejardin, 2006). The control of this process is dependent upon the presence of NF-kB inducing kinase (MAP3K14/NIK), a protein that is normally undetectable as it is co-translationally degraded in a TNF receptor associated factor 3 (TRAF3) dependent manner (Liao et al., 2004). Consistently, Traf3−/− mice are characterized by constitutive processing of NFKB2 (He et al., 2006a). Another TNF receptor associated factor, TRAF2, may also play a role in the control of NIK function as a conditional Traf2 knockout in B-cells results in constitutive processing of NFKB2 (Grech et al., 2004).
The importance of the non-canonical NF-kB pathway in B-cell development has been established in a number of model systems (Sen, 2006). First, the classical alymphoplasia mouse phenotype is caused by a missense mutation of Nik (Nikaly/aly) that prevents the induction of NFKB2 processing (Shinkura et al., 1999). Additionally, Nfkb2−/− mice display a reduced B cell compartment, poor germinal center development but a normal T cell compartment (Caamano et al., 1998; Franzoso et al., 1998). Conversely, transgenic mice expressing constitutively active isoforms of NFKB2, analogous to the ones identified in a variety of lymphoid malignancies (Neri et al., 1991), have an expansion of their B cell population (Ciana et al., 1997; Ishikawa et al., 1997).
In addition to their role in B cell homeostasis, the NF-kB pathways have been implicated in a wide variety of tumors, however, only a limited number of activating mutations have been reported (Courtois and Gilmore, 2006; Karin, 2006). Using a combined strategy of high-resolution array-based comparative genomic hybridization (aCGH) and gene expression profiling (GEP) we have identified abnormalities in a number of genes predicted to result in dysregulation of primarily the non-canonical NF-kB pathway. These mutations provide a genetic basis for the dysregulation of NF-kB in some cases of MM and their genetic selection highlights the paramount importance of the NF-kB pathway in this disease.
Results
aCGH identifies frequent bi-allelic deletions of NF-kB regulatory genes
In order to identify genetic abnormalities in MM we screened 62 patient samples and 46 human myeloma cell lines (HMCLs) originating from 42 individuals on a high density oligonucleotide aCGH platform with 42 896 features at an average genomic interval of ~35 kb (Barrett et al., 2004). We took a reductionist approach to the analysis of the aCGH data by focusing our analysis on bi-allelic/homozygous deletion events, which are intrinsically limited to small genomic regions but are likely to harbor tumor suppressor genes (Figure S1). To ensure that the observed bi-allelic deletions represented genuine events and not noise in the data or large common nucleotide variations (CNV)(Khaja et al., 2006; Redon et al., 2006), we first applied a circular binary segmentation algorithm (CBS) to the data (Olshen et al., 2004). The CBS algorithm provides a very stringent means of identifying bi-allelic deletions as it segregates chromosomal locations into regions with significant adjacent mean copy number changes. Although this reduced the resolution of the analysis from ~35kb to ~70kb, it significantly limits the number of false positive events. This analysis scheme identified 13 bi-allelic deletion events in 11 patients, encompassing 43 genes, and 80 bi-allelic deletions in 36 HMCLs, encompassing 148 genes, that do not represent physiological rearrangements of immunoglobulin loci or aberrations within previously characterized CNV regions (Table S1).
The only recurrent bi-allelic deletion identified by aCGH in our patient samples occurred at 14q32 and encompassed three potential target genes; TRAF3, AMN, and CDC42BPB (Figure 1 & S2). Furthermore, this abnormality was one of only two identified in both our patient and HMCL datasets. We confirmed the bi-allelic deletions by cIg-FISH and the minimally deleted region (MDR) was mapped by PCR in the two HMCLs with bi-allelic deletions to a genomic region of <48.8 kb using sequence tagged site (STS) and fine mapping primer pairs (Figure 1). This fine mapping approach eliminated CDC42BPB as a target gene of the deletion and thus only TRAF3 or AMN remained as potential target genes. The remaining bi-allelic deletion identified in both of our datasets occurred at 11q22. This deletion was often quite large, ranging from 0.14–4.22 Mb in size, however, the MDR defined by aCGH was <109.05 kb in size and encompassed only 2 genes; BIRC2/cIAP1, and BIRC3/cIAP2 (Figure 1). As expected, the presence of a bi-allelic deletion correlated with reduced expression of the predicted target gene (Figure S3).
Figure 1. High Resolution aCGH Identifies Bi-Allelic Deletions of NFKB Regulatory Genes.

A) The bi-allelic deletions (TRAF3, cIAP1 or cIAP2, CYLD, TRAF2) predicted to target NF-kB regulatory genes identified in our 62 MM patient and 46 HMCL aCGH cohorts are shown. All of the maps except the TRAF3 map, which is drawn to relative scale so that the exon structure can be shown, are drawn to scale. Black arrows above each gene represent the direction of transcription. For TRAF3 the direction of transcription follows the numerical exon order and light grey exon regions indicate coding region. Each bi-allelic deletion is shown below the respective genomic map. Solid grey bars indicate regions of bi-allelic deletion, solid grey lines indicated regions of bi-allelic deletion mapped by PCR, and dashed grey lines indicate the region containing the breakpoint. The position and the copy number prediction of the aCGH probes contain on the microarray are indicated by red (1 or more copies) or green (0 copies) dots. The position of probes used for cIg-FISH are shown below each genomic map. Dashed vertical red lines indicate the MDR identified by aCGH or PCR mapping. B) Representative images from the cIg-FISH validation of the aCGH findings are shown. Myeloma cells are identified by the blue cytoplasmic stain, which indicates the presence of either cytoplasmic kappa or lambda light chains. In the cIAP1/2 and TRAF2 panels a macrophage (kidney shaped nucleus) is shown that takes up the stain unspecifically. In the TRAF2 panel one of the three plasma cells shown still retains a copy of TRAF2, fitting the observed frequency of bi-allelic TRAF2 deletions observed in this patient sample.
Interestingly, inactivation of TRAF3, cIAP1/2, and CYLD appears to be a common event in MM, as we identified a number of potential bi-allelic deletions in the publicly available dataset from Carrasco et al., based on a correlation between the aCGH copy number and the expression level (Figure S4)(Carrasco et al., 2006). Combining these two patient datasets did not help to further refine the MDR encompassing TRAF3 or cIAP1/2, however, it did refine the MDR of the bi-allelic deletion we observed at 16q12 to a 72.05 kb region, which includes two genes, CARD15 and CYLD (Figure S4).
Interestingly, 5/13 (38.5%) of the bi-allelic deletion events identified within our patient cohort contained potential target genes (TRAF2, TRAF3, cIAP1/2, and CYLD) associated with the regulation of the NF-kB signaling pathways. Based on these observations we developed the working hypothesis that the frequent dysregulation of NF-kB signaling observed in MM is often the result of genetic abnormalities within the tumor cells.
Combined analysis of GEP and aCGH datasets identifies additional abnormalities within regulators of the NF-kB signaling pathways
Our primary analysis of the aCGH datasets identified inactivation of 5 different genes involved in the NF-kB signaling pathways. To build on these observations we combined our aCGH and previously generated GEP datasets to identify additional abnormalities within genes involved in the NF-kB signaling pathways. A screen of our 125 patient and 44 HMCL GEP cohorts for spiked expression of genes involved in the regulation of NF-kB signaling identified over-expression of MAP3K14/NIK, LTBR, CD40, TNFRSF13B/TACI, and NFKB1 (Figure 2 & S3).
Figure 2. NFKB Related Genes with spiked Expression correlate with genetic Abnormalities.
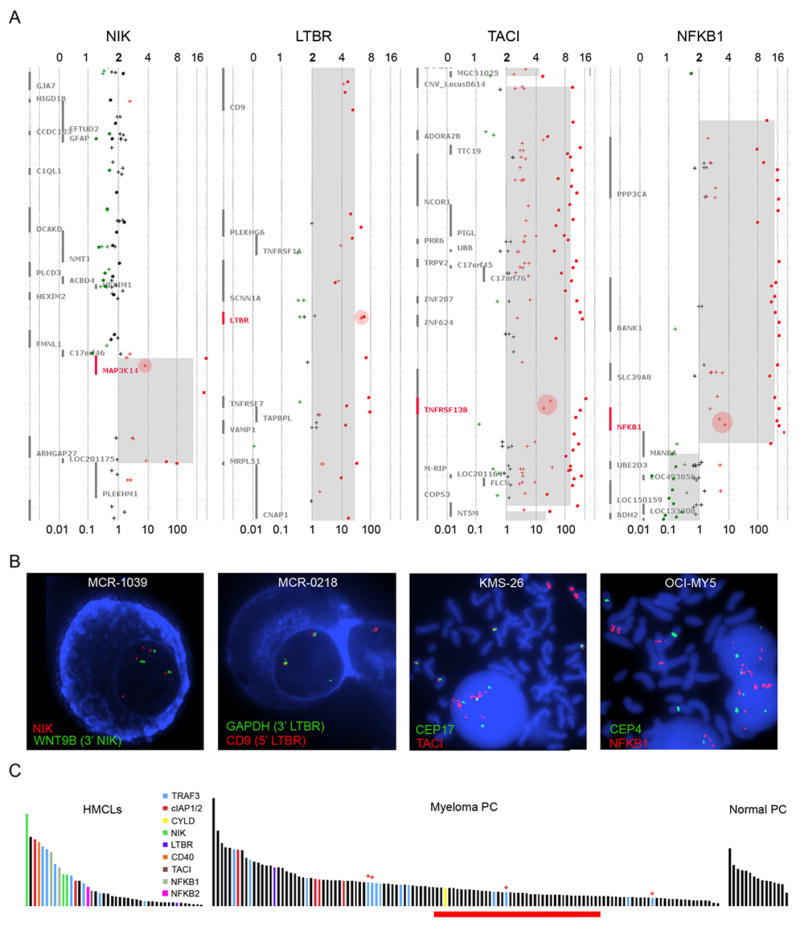
A) Representative plots indicating the regional copy number abnormality and corresponding gene expression level are shown. The gene of interest is highlighted in red on each plot. The predicted number of gene copies is indicated on the top of each panel and the median normalized expression level is shown at the bottom. Each aCGH probe is represented by a dot and the color of each dot corresponds to the log2 ratio of the probe; black dots, normal copy number (log2 ratio -0.25-0.25); green dots (log2 ratio <-0.25); red dots (log2 ratio >0.25). The light grey region represents the mean copy number change identified by the CBS algorithm. Crosses indicate the gene expression probes for each gene and the log10 transformed expression levels are color coded to match the aCGH data. Pink circles highlight the expression probes corresponding to the gene of interest. B) Representative cIg-FISH images are shown that document the rearrangements of NIK and LTBR in MM patients, the duplication and insertion of CD40 into the Ig lambda locus, and the HSR containing TACI and NFKB1. C) Histogram plots of the NFKB index from normal PC, myeloma PC, and HMCL are shown and indicate the correlation between the NFKB index and the presence of an abnormality in the NF-kB pathways. The red bar below the index corresponds to the predicted transition point between processed versus unprocessed NFKB2. The red asterisks identify samples with questionable TRAF3 inactivation due to either a heterozygous mutation call by sequencing or a bi-allelic deletion in less than 25% of the plasma cells.
We identified the dysregulated expression of NIK in two HMCLs, L363 and EJM, that have previously been reported to over-express NIK by either a t(17;22) translocation or amplification, respectively (Annunziata et al., 2006). Because of these observations we examined the aCGH datasets for abnormalities of the NIK locus. We confirmed the amplification in EJM, and identified unbalanced rearrangements of NIK in one HMCL, JJN-3, and one patient, which were confirmed by cIg-FISH (Figure 2 & S5).
We noted spiked expression of LTBR and CD40 in both patient and HMCL cohorts. A >40-fold increase in the expression of LTBR was observed in one patient and in the HMCL FR4. For FR4, aCGH showed a significant amplification of the entire 12p chromosome arm, which was confirmed by FISH (Figures 2 & S6). Interestingly, LTBR is one of only six genes on the entire arm with a >40-fold increase in expression. For the one patient with spiked LTBR expression aCGH data was not available; however, FISH identified a single copy gain of the LTBR locus that is inserted at an unknown location (Figure S6). Within the surrounding region only LTBR and VAMP1 are over-expressed in this patient (Figure S6). For CD40, we identified one HMCL and two patient samples with spiked expression. By aCGH the HMCL XG-2 contains a discrete two-copy gain of the CD40 locus, which was confirmed by FISH and shown to be associated with the Ig-lambda locus (Figures S7). In the two patients with spiked CD40 expression, aCGH data were not available and cIg-FISH did not detect a copy number variability or rearrangement of CD40.
We also detected the spiked expression of TNFRSF13B/TACI in the HMCL KMS-26 and NFKB1 in OCI-MY5 and KMM-1. In both KMS-26 and OCI-MY5 the over-expression of TACI and NFKB1 were mirrored by the presence of homologous staining regions containing the respective genes (Figure 2). In KMM-1, which had a 19-fold increase in the expression of NFKB1, a detectable alteration by aCGH was not identified. However, by FISH an expression cloud was associated with a single NFKB1 allele that disappeared following RNase treatment (Figure S8), suggesting that an unknown mechanism is driving the over-expression of NFKB1 from a single allele in this HMCL.
Finally, we were able to correlate the presence of these diverse mutations with increased NF-kB transcriptional activity (Figure 2). The level of NF-kB mediated transcriptional activity was determined based on a gene expression index developed from the HMCLs. We used an ANOVA test using the error estimate from a cross-gene error model with multiple testing correction to identify 6 probesets (p<0.05) differentially expressed between the HMCLs with identified mutations compared to those without identified NFKB pathway mutations. We chose to remove two probe sets corresponding to HLA class II genes as their expression in patient samples may also derive from contaminating normal monocyte/macrophages and B-cells. The mean expression level of the remaining 4 probesets corresponding to CD74, IL2RG, and TNFAIP3 (2x) was used to represent the NFKB index. NF-kB transcriptional activity appeared to be high in all normal bone marrow plasma cells, likely due to stimulation from the surrounding microenvironment, but was variable in MM patients, and HMCL. Clearly, the presence of mutations in the NF-kB pathways was associated with a higher level of NF-kB transcription activity in the HMCLs. This correlation was also observed in our MM patients, albeit to a lesser degree, but the correlation is also evident in other datasets (Figure S4). The reduced correlation between the NF-kB index and mutations seen in the patient dataset may in part be due to the limitations inherent in the analysis of clinical material and to other sources of NF-kB activation originating from the bone marrow. However, in both datasets the association of these mutations with NF-kB transcriptional activity is not likely to result from chance alone as a hypergeometric distrubution test, a statistical test commonly used in enrichment analysis, returned p-values of 0.009 and 0.04 respectively.
FISH validation of genetic abnormalities within the NF-kB signaling pathways
To determine the frequency of TRAF3 deletions we blindly screened a large cohort of MM patients by cIg-FISH, including 60 patients initially screened by aCGH and 98 additional patients. We identified 6 (3.8%) patients with bi-allelic TRAF3 deletions and 19 (12.0%) patients with a single TRAF3 allele (Table 1). There was almost complete concordance between the FISH and aCGH results with the exception of two patients in whom the presence of the bi-allelic deletion was heterogeneous by FISH and below the detection limit of aCGH. However, in both these cases, aCGH demonstrated a clear loss of chromosome 14, which suggests that the bi-allelic deletions seen by FISH occurred after the q-arms of chromosome 14 were lost.
Table 1.
Frequency of Genomic Deletions within NF-kB Related Genes
| Gene | Sample | Assay | n | Number of Gene Copies | ||
|---|---|---|---|---|---|---|
| 2N/3N/4N | 1N | ON | ||||
| TRAF3 | HMCL | aCGH | 42* | 29 (69.1) | 10 (23.8) | 3 (7.1) |
| Patients | FISH | 158 | 133 (84.2) | 19 (12.0) | 6 (3.8)‡ | |
| aCGH | 62 | 54 (87.1) | 6 (9.7)‡ | 2 (3.2) | ||
| Total | 160 | 134 (83.7) | 19 (11.9) | 7 (4.4) | ||
| TRAF2 | HMCL | aCGH | 42 | 35 | 7 | 0 |
| Patients | FISHΠ | 7 | 6 | 0 | 1 | |
| aCGH | 62 | 60 (96.8) | 1 (1.6) | 1 (1.6) | ||
| cIAP1/2 | HMCL | aCGH | 43† | 36 | 7 | 3 |
| Patients | FISH∫ | 15 | 11 | 0 | 4 | |
| aCGH | 62 | 60 | 1 | 1 | ||
| CYLD | HMCL | aCGH | 43§ | 34 (79.1) | 9 (20.9) | 0 |
| Patients | FISH£ | 30 | 13 | 15 | 2 | |
| aCGH | 62 | 50 (80.7) | 11 (17.7) | 1 (1.6) | ||
A total of 46 HMCL were screened by aCGH, however, the 46 HMCL only represent 42 individuals because some of the lines originate from the same individual. The following HMCL were screened: ANBL-6, Delta47, EJM, FLAM-76, FR4, H1112, INA-6, JIM3, JJN-3, JK-6L Schon, Karpas620, KHM-11, KHM-1B, KMM-1, KMS-11, KMS-12BM, KMS-12PE, KMS-18, KMS-20, KMS-26, KMS-28BM, KMS-28PE, KMS-34, L363, LP-1, MM.1R, MM.1S, MM-M1, NCI-H929, OCI-MY1, OCI-MY5, OCI-MY7, OPM-1, OPM-2, PE-1, PE-2, RPMI-8226, SACHI, SKMM-1, SKMM-2, U266, UTMC-2, XG-1, XG-2, XG-6, XG-7
Although the aCGH defined copy number of the TRAF3 locus in KMS-11 is 1N it has been placed in the ON category as a bi-allelic deletion of exon 7, an unrepresented region on the aCGH chip, was discovered during exon resequencing.
Two patients are differentially represented in the 1N and ON categories between the FISH and aCGH result categories. In both cases the bi-allelic deletion of TRAF3 was seen in a sub-population of the light chain selected plasma cells while the remaining populations show only a single copy of TRAF3. Because of this mixed population both patients are characterized as 1N by aCGH.
The total number of HMCL is noted as 43 as KMS-28PE shows a bi-allelic deletion of cIAP1 and cIAP2, while the sister HMCL KMS-28BM, derived 1 month later (Takemi Otsuki, Pers. Comm.), shows a 1N copy number
The total number of HMCL is noted as 43 as KMS-12BM shows a single copy of CYLD while the sister HMCL KMS-12PE, derived 2 months earlier (Takemi Otsuki, Pers. Comm.), shows the normal 2N copy number.
FISH confirmed the bi-allelic TRAF2 deletion observed in one patient by aCGH.
We screened a panel of patients for cIAP1 and cIAP2 abnormalities based on their low expression of cIAP1 as detected by GEP, or the prediction of a 1N copy number by aCGH. This panel includes 13 patients who were not screened by aCGH. FISH confirmed the bi-allelic deletion observed in one patient by aCGH.
We screened a panel of patients for CYLD abnormalities based on their low expression of CYLD, as detected by GEP, or the prediction of a 1N copy number by aCGH. This panel includes 9 patients for whom aCGH data is not available. FISH confirmed the bi-allelic CYLD deletion observed in one patient by aCGH.
To systematically screen the limited patient material for deletions of TRAF2, cIAP1/2 and CYLD by cIg-FISH we selected patients for screening based on either 1N copy number of these genes, as determined by aCGH, or a median normalized expression below 0.4. This process identified three additional patients with cIAP1/2 deletions and one additional patient with a CYLD bi-allelic deletion (Table 1). No additional cases were identified with TRAF2 deletions. In summary, we identified bi-allelic deletions of TRAF2, TRAF3, CYLD or cIAP1/2 in 14/167 patients, and of TRAF3, CYLD or cIAP1/2 in 10/67 in the Carrasco dataset. We believe that the cumulative incidence of these abnormalities is under-estimated as only TRAF3 was studied in the majority of patients due to the limited availability of patient material.
Sequencing identifies mutations of TRAF3 and CYLD
To determine the overall rate of inactivation of the identified target genes we performed bi-directional sequencing of the coding regions of these genes. The individual coding exons and flanking mRNA splicing sites of TRAF3 were sequenced in 62 patients and in the entire panel of 46 HMCLs. Through this process, we found a third HMCL, KMS-11, with a bi-allelic deletion of TRAF3 originating from two independent deletions overlapping at exon 7 (data not shown). In the remaining HMCLs we found four independent mutations including three frameshifts and one in-frame deletion, which are predicted to inactivate TRAF3 (Figure 3, Tables 2 & S2). All HMCL, except the three HMCLs with bi-allelic deletions, expressed detectable full-length TRAF3 mRNA transcripts and no discernable transcript length variations, suggestive of unknown alternative splicing events, were observed.
Figure 3. Sequencing identifies mutations in the coding region of TRAF3.
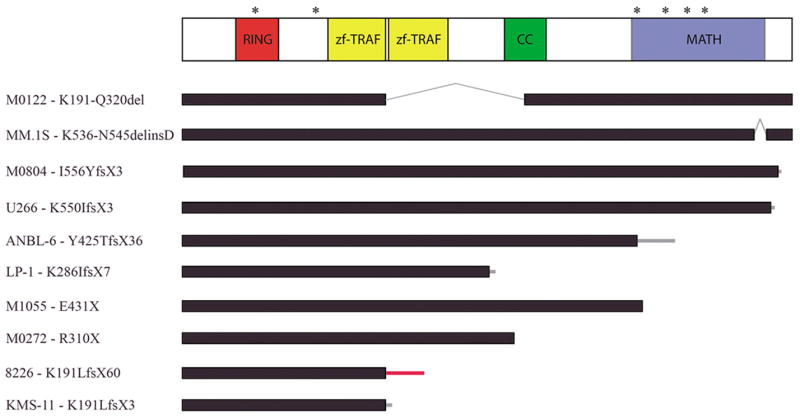
A scale diagram of the TRAF3 polypeptide and the known protein domains identified by the SMART prediction tool are shown. Asterisks above the diagram note the position of the identified missense mutations (H70Y, R118W, Q430R, G462A, G480E, F490C) while the nonsense, frameshift and deletion mutations and the associated polypeptide effects are shown below as black bars. Black bars represent normal coding regions, grey lines indicate deleted regions, and thin bars represent the position of a frameshift and the length of translation before a stop codon is encountered. The thin grey bars indicated the translation of sequence originating within the TRAF3 locus while red bars indicate the translation of regions outside of the TRAF3 locus.
Table 2.
Inactivation of NF-kB Pathway Genes
| Gene | Sample | Samples Sequenced | Inactivating Mutations | Inactivation | ||
|---|---|---|---|---|---|---|
| 2N/3N/4N | 1N | ON | ||||
| TRAF3 | HMCL | 41† | 1/29 (3.4) | 4/10 (40.0) | 0/3 | 7/42 (16.7) |
| Patients | 62* | 1/43 (2.3) | 8/16 (50.0) | 1/3 | 12/62 (19.4)‡ | |
| CYLD | HMCL | 16 | 0/7 | 0/9 | 0/16 | |
| Patients | 11 | 0/6 | 1/5 | 1/11 | ||
An additional 15 patients were partially sequenced for TRAF3, cIAP1, cIAP2, and CYLD abnormalities at the University of Toronto. A single patient harbored a heterozygous nonsense mutation (c.900C>T, Q183X) of TRAF3. In addition to the observed mutation a number of previously undocumented SNP were identified in TRAF3, CYLD, cIAP1, and cIAP2.
OCI-MY1 was not sequenced because the entire TRAF3 locus is deleted
An initial panel of 49 unselected patients was screened for TRAF3 coding mutations, in which 3 mutations were identified. Subsequently, a selected population of 13 patients with a single TRAF3 allele, or mixed population of 1N and ON defined by FISH, was screened for TRAF3 mutations if template material was available for cDNA or DNA based sequencing approaches and this identified 7 additional mutations.
The expected frequency of TRAF3 inactivation by mutation or bi-allelic deletion after a 1N selection bias correction is ~12.3%.
Correction Calculation = (Bi-allelic Frequency + (1N Frequency × 1N Mutation Frequency) + (2N/3N/4N Frequency × 2N/3N/4N Mutation Frequency)
We sequenced 49 randomly selected patient samples for TRAF3 and identified 3 different mutations (one nonsense and two missense). Subsequently, TRAF3 sequencing was extended to an additional 13 patients in whom LOH of the TRAF3 locus had been characterized by aCGH or cIg-FISH, and DNA or RNA was available for sequencing. This process identified 7 additional abnormalities within the coding region of TRAF3, including one nonsense, one frameshift, and four missense mutations along with one large deletion (Table 2 & S2). All of the missense mutations occur at amino acids conserved throughout vertebrate evolution (Figure S9), suggesting their functional relevance. Surprisingly, one of the patients with a subpopulation of cells with a bi-allelic deletion of TRAF3, MCR-0319, also had a missense mutation, which inactivates the RING domain of TRAF3 (Figures 3 & S9). A domain recently shown to be essential for the negative regulation of NIK by TRAF3 (He et al., 2006a). Altogether, TRAF3 is inactivated in 17% of HMCL and approximately 12.3% of MM patients by either bi-allelic deletion or by LOH associated with an inactivating mutation.
We sequenced the coding exons of CYLD in 11 patients with either low levels of CYLD expression or LOH and identified a single nonsense mutation, S265X, in one patient sample with LOH (Table 2 & S2). No mutations were identified in HMCLs with LOH of CYLD (Table 2). In the HMCL JK-6L, we identified a frameshift mutation in NFKB2, which was investigated based on the presence of an abnormal immunoblot band. In the only patient with LOH of the cIAP1/2 locus we did not detect an abnormality within the coding region of cIAP1 or cIAP2.
In summary, our comprehensive genetic screen identified a promiscuous array of genetic alterations that are predicted to result in constitutive activation of NF-kB signaling. When all of the bi-allelic deletion events, mutations, and gene rearrangements of TRAF2, TRAF3, cIAP1/2, CYLD, NIK, LTBR, CD40, TACI, NFKB2 and NFKB1 are combined, at least 28/167 (17%) MM patients and 19/46 (41.3%) HMCL have an abnormality that leads to the dysregulation of NF-kB signaling.
Genetic Alterations of TRAF3, BIRC2, BIRC3, CD40, NIK, TACI, LTBR correlate with the processing of NFKB2
To determine if the genetic aberrations we identified were associated with constitutive processing of NFKB2, we screened the panel of HMCLs for the processing of p100 to p52 (Figure 4 & Table S3). We identified constitutive NFKB2 processing in HMCLs with abnormalities of TRAF3, cIAP1/2, NIK, CD40, LTBR, and TACI. As expected, the HMCLs that over-express NIK or have TRAF3 inactivating abnormalities had constitutive NFKB2 processing while HMCL over-expressing NFKB1, OCI-MY5 and KMM-1, did not have detectable NFKB2 processing. The constitutive processing of NFKB2 from p100 into p52 correlates with both a high NFKB index and the detection of the normally cytoplasmic NFKB2 in the nucleus (Figure 4)(Table S3). In addition, the HMCLs with inactivation of TRAF3 and rearrangements of NIK were associated with detectable levels of NIK protein. Surprisingly, we also found high levels of NIK and constitutive processing of NFKB2 in HMCLs with cIAP1/2 bi-allelic deletions (Figure 4). The constitutive processing of NFKB2 seen in these HMCLs must be linked to the absence of cIAP1 and/or cIAP2 as the isogenic HMCL pair (KMS-28BM/PE), which were independently derived from the bone marrow or the pleural effusion of the same patient and share virtually identical genomes except for their difference in the presence (KMS-28BM) or absence (KMS-28PE) of cIAP1/2 also process NFKB2 differently. In this pair, the absence of cIAP1/2 is associated with increased levels of NIK protein and constitutive NFKB2 processing (Figure 4). As all of our bi-allelic deletions encompassed both cIAP1 and cIAP2 and we did not identify a mutation in either gene, we cannot determine if the loss of one or both is associated with NFKB2 processing.
Figure 4. Mutations in the non-canonical NF-kB pathway correlate with processing of NFKB2 and the presence of NFKB2 in the nucleus.

A) Immunoblots for the proteins of interest from the indicated HMCLs are shown. The three vertical panels are separated into a TRAF3, cIAP1, and NIK panel plus additional HMCL when possible. For the NFKB2, TRAF3, and NIK blots L363 and OPM-1 were included on the ends of each blot to serve as positive and negative controls and to ensure equivalent exposure between each panel. The cIAP1 blot for L363 has been taken from another gel as we had included KMS-18 as a negative control in all of the blot panels and this prevented L363 from being included. In all instances the bi-allelic deletion of TRAF3 or cIAP1 corresponds with the complete absence of a detectable protein product. B) The association between processing of NFKB2 and nuclear localization of NFKB2 was confirmed by immunofluorescence of HMCL without NFKB2 processing (OCI-MY5) compared to those with constitutive NFKB2 processing (L363, NIK overexpression; OCI-MY1, TRAF3 bi-allelic deletion; and U266, homozygous TRAF3 mutation). C) An NFKB2 immunoblot confirms the existence of NFKB2 processing in MM patient samples and shows a correlation between the processing ratio (left-right; positive, negative, positive, negative) and NFKB index (left-right; 1.07, 0.45, 1.26, 0.56). D) Immunohistochemistry of patients with a low (MCR-0459) or high (MCR-0687) NFKB index along with a patient with a TRAF3 bi-allelic deletion (MCR-0387) confirms the association between a high NFKB index or TRAF3 mutation and nuclear NFKB2. The images are representative of the 5 patient samples tested from each category. All core biopsies but one with sufficient plasma cells for scoring correlated with expectations of nuclear NFKB2 in TRAF3 mutants or samples with high NFKB index while samples with no know mutation and a low NFKB index had cytoplasmic NFKB2.
Surprisingly, 2 of 3 HMCLs, L363 and EJM, with NIK abnormalities did not have detectable TRAF3 protein. However, the third cell line, JJN-3, which has a NIK rearrangement deleting the TRAF3 interaction domain (Pers. Comm. Michael Kuehl), had high levels of TRAF3 protein. Although it is known that TRAF3 regulates NIK protein stability, these two observations suggest that a direct NIK-TRAF3 interaction may also regulate TRAF3 protein levels. Furthermore, the high levels of TRAF3 in the cell lines lacking cIAP1/2 suggest this may occur in a cIAP1/2 dependent manner.
In conclusion, the majority of the genetic abnormalities we identified in our cohort of MM patients and HMCLs have highlighted a multi-faceted mechanism that constitutively activates the non-canonical NF-kB pathway. The selection of so many mutations in a single pathway highlights its importance in myelomagenesis and identifies a potential tumor specific vulnerability.
Characterization of TRAF3 as a Tumor Suppressor Gene
Since TRAF3 inactivation represented the most common abnormality we identified in the non-canonical NF-kB pathway in MM, we investigated its potential role as a tumor suppressor. We constructed an adenovirus capable of high efficiency infection, which expresses wild-type TRAF3 and used it to reintroduce TRAF3wt into the HMCLs with TRAF3 abnormalities. Based on the well-documented role of TRAF3 as a negative regulator of NFKB2 processing we assayed its effects on p100 processing. In all of the HMCLs with TRAF3 abnormalities the reintroduction of TRAF3wt was associated with an inhibition of NFKB2 processing from p100 to p52 (Figure 5). In addition, over-expression of TRAF3 in the HMCL L363, which over-expresses NIK due to an IgL translocation, leads to a marked inhibition of NFKB2 processing.
Figure 5. TRAF3 reintroduction inhibits NFKB2 processing and causes a growth arrest.

The effects on HMCLs infected with an adenovirus expressing either EGFP and TRAF3WT or EGFP alone are shown A) Immunoblot showing the inhibition of p100 processing 48 hrs post infection. B) Effects of TRAF3WT expression on MTT defined cell growth. Infected cells were seeded 24 hrs post infection and the effects on cell growth we compared to control infections at 72 hrs and 96 hrs post-infection. Cells were seeded at specific densities to ensure that the cells infected with the EGFP expressing control virus remained in the exponential phase of the growth curve at both time measurements. Error bars represent the standard deviation C) Effect of TRAF3WT expression on the cell cycle status of selected HMCL 48 hrs post infection. The scale for each plot has been normalized for each HMCL. D) TRAF3WT reintroduction is associated with a significant increase in apoptosis as measured by Annexin-V staining.
The involvement of TRAF3 in the regulation of NIK stability and the subsequent p100 processing is well documented, however, a role in oncogenesis has never been reported. We hypothesized that the inactivation of TRAF3 would provide cells with a selective growth advantage. As we predicted, the re-introduction of TRAF3wt caused a significant growth delay in 4/5 HMCL associated with G0/G1 cell cycle arrest and a significant increase in apoptosis (Figure 5). The control HMCLs, OCI-MY5 and OPM-1, that do not have constitutive processing of NFKB2 were not affected by TRAF3 over-expression.
Inactivation of TRAF3 is associated with poor response to dexamethasone and a good response to proteasome inhibitors
Proteasome inhibitors are a novel class of anti-cancer agents with remarkable clinical activity in MM (Richardson et al., 2003). Although they presumably have multiple mechanisms of action, the principally defined mechanism has been postulated to be the inhibition of NF-kB signaling. We hypothesized that MM with constitutive activation of the non-canonical pathway would be particularly sensitive to proteasome inhibition. Unfortunately we could not test this hypothesis in the HMCLs, which are uniformly sensitive to proteasome inhibitors and differ from patients in a number of ways, most notably proliferation. Instead we examined a GEP dataset of relapsed/refractory MM patients randomly assigned to treatment with either dexamethasone or the proteasome inhibitor bortezomib (Mulligan et al., 2006). As we could not screen these patient samples for mutations, we used an ROC analysis of our patient dataset to identify a median normalized expression cutoff of 0.6 as the best crossover point between sensitivity and specificity and used this value as a surrogate means of identifying patients who likely have TRAF3 abnormalities. In our dataset 17/25 patients with TRAF3 expression below 0.6 lack functional TRAF3. Using this cutoff, 39/193 (20%) patients likely have a TRAF3 abnormality, a rate similar to that observed in our cohort. In this clinical trial dataset 2/20 (10%) patients with low levels of TRAF3 responded to dexamethasone whereas 17/19 (89%) responded to bortezomib. This was accompanied by a prolongation of progression-free-survival (PFS) from 83 to 193 days (p<0.0001). For patients with levels of TRAF3 above 0.6, 15/50 (30%) responded to dexamethasone and 42/104 (40%) to bortezomib, with no difference in PFS (140 vs 145 days)(Figure S10). These results suggest that constitutive activation of the non-canonical NF-kB pathway by inactivation of TRAF3 is associated with dexamethasone resistance and proteasome inhibitor sensitivity. The resistance to dexamethasone was unexpected but may relate in part to the fact that one of the mechanisms of action of glucocorticoids targets only the canonical NFkB pathway. It has been reported that hormone-bound glucocorticoid receptor is recruited to promoters by binding relA-containing dimers, thereby disrupting relA’s interaction with certain co-activators(Hoffmann and Baltimore, 2006). One would anticipate that this mechanism would be of little consequence in patients with constitutive activation of the non-canonical NF-kB pathway. Using the top and bottom third of the NFKB index to discriminate patients shows that patients in the high index group have a better response rate to bortezomib, although no difference in PFS. These results suggest that bortezomib may be most effective in patients with constitutive non-canonical NF-kB activation (as seen in patients with inactivation of TRAF3).
Discussion
Our integrated analysis of high-resolution aCGH and GEP identified a plethora of genetic abnormalities resulting in the activation of the NF-kB signaling pathways. We observed over-expression and/or gain-of-function mutations in NIK, NFKB2, NFKB1, CD40, LTBR, and TACI, which are all positive regulators of NF-kB signaling. Furthermore, we identified inactivating abnormalities of TRAF3, cIAP1/cIAP2, CYLD, and TRAF2, which are negative regulators of the NF-kB pathways. Importantly, these observations provide valuable insight into the significant contribution of the non-canonical NF-kB pathway in the pathogenesis of MM. In the course of this study we have made several compelling observations. First, TRAF3 is a tumor suppressor gene that is inactivated at a frequency greater than any other known tumor suppressor in MM. Furthermore, its reduced expression correlates with dexamethasone resistance and bortezomib sensitivity. Second, inactivation of cIAP1 and/or cIAP2 by bi-allelic deletion is associated with an activation of the non-canonical NF-kB pathway. Third, we found an inactivating mutation in CYLD associated with LOH, suggesting that CYLD, the causative gene in cylindromatosis, may play a role in the development of MM.
To date, the dysregulation of NF-kB signaling observed in MM has largely been linked to ligand-dependent interactions occurring in the bone marrow. We have found that plasma cells isolated from the bone marrow of healthy individuals have an elevated NF-kB transcriptional index, suggesting that activation of NF-kB is a physiological feature of these cells. The ligand dependent NF-kB stimulation of normal plasma cells likely serves to regulate the growth and survival of these cells within the confines of the bone marrow compartment. We propose that the acquisition of mutations reported here, results in the accumulation of malignant plasma cells beyond this physiological control. Our results suggest a mechanism by which MM cells can overcome these limitations through the acquisition of mutations that result in constitutive and ligand-independent activation of the non-canonical NF-kB pathway.
The regulation of the non-canonical NF-kB pathway is absolutely dependent on the presence of NIK that, although constitutively transcribed, is normally undetectable in the absence of receptor engagement (Qing et al., 2005), which sequesters the normally cytoplasmic localized TRAF2, TRAF3, and cIAP1/cIAP2 proteins to the plasma membrane (Fotin-Mleczek et al., 2004). Based on our observations we predict the existence of a cytoplasmic complex regulating the co-translational degradation of NIK, which relies on the presence of TRAF2, TRAF3, cIAP1 and/or cIAP2 (Figure 6). We hypothesize a mechanism in which, in the absence of stimuli, cytoplasmic TRAF3 scavenges the cytoplasm for NIK, which is then recruited to complexes containing TRAF2, cIAP1, and cIAP2 for degradation. Upon receptor engagement, these regulators are sequestered to the plasma membrane, leading to NIK stabilization. We believe that any mutation affecting the binding of these regulators to each other and to NIK would result in NIK stabilization. In conclusion, the genetic abnormalities we have identified are expected to contribute to the constitutive processing of NFKB2 by either inhibiting the function of the NIK regulatory complex (TRAF2, TRAF3, cIAP1 and/or cIAP2 abnormalities), bypassing or overwhelming the NIK regulatory complex (NIK or NFKB2 abnormalities), or by recapitulating the natural process that inactivates the NIK regulatory complex by recruiting the various constituents to the plasma membrane (LTBR, TACI, and CD40 over-expression).
Figure 6. Proposed Model Indicating the Effects of the Identified Abnormalities on the Non-Canonical NF-kB Pathway.

The genes with inactivating abnormalities; TRAF2, TRAF3, cIAP1, cIAP2, and CYLD, are indicated by green objects while the genes with activating abnormalities; LTBR, TACI, CD40, NIK, and NFKB2, are indicated by red objects. Based on our observations and those from other groups we propose the existence of a cytoplasmic NIK regulatory complex, shown in the light red hexagon, that is dependent on TRAF2, TRAF3, cIAP1 and/or cIAP2.
While TRAF3 is a recognized regulator of the non-canonical NF-kB pathway, it has never been implicated in a malignant process. In this study we have found that TRAF3 is inactivated at a frequency greater than that of any other tumor suppressor in MM. Given this frequency, we suspect that other malignancies might harbor TRAF3 abnormalities. In fact, 21 of 83 cell lines derived from a variety of malignancies lacked detectable TRAF3 by immunohistochemistry (Zapata et al., 2000).
In conclusion, we have identified a promiscuous array of mutations that result in the constitutive activation of the non-canonical NF-kB pathway in approximately 20% of MM patients. The remarkable activity of bortezomib in patients with inactivation of TRAF3 suggests that one of its most important mechanisms of action in MM is the inhibition of the non-canonical NF-kB pathway. Therefore, we believe that the specific targeting of the direct NFKB2 regulators NIK and IKKα may be particularly effective in MM treatment.
Materials and Methods
Sample Preparation
Bone marrow aspirates were collected from 167 MM patients (28 SMM and 139 overt MM). All patients provided written informed consent approving the use of their samples under Institutional Review Board approval. The BM aspirates were treated with ACK lysis buffer to remove red blood cells and plasma cells were isolated using immuno-magnetic sorting on a Miltenyi AutoMacs or StemCell Robocept with anti-CD138 antibodies. We enumerated 100 nucleated cells for kappa and lambda staining to determined the purity of the CD138 sorts and found that the mean plasma cell purity (kappa & lambda) was 95.5% (Median 96.0, range 69–100%) while the mean clonal purity (clinical light chain) was 92.7% (Median 95.0, range 54–100). Isolated cells were suspended in TRIzol (Invitrogen) and stored at −80°C for long-term storage. Nucleic acids were isolated from TRIzol following the protocol supplied by the manufacturer. The RNA used for gene expression profiling was cleaned up using the Qiagen RNeasy kit, while the DNA used for aCGH was cleaned up by phenol-chloroform extractions after RNase and proteinase K treatments. The HMCLs were maintained as previously described(Bergsagel et al., 1996).
Microarray experiments
GEP of the patient samples was performed on U133A genechips following the manufacturer's suggested protocol, while the 46 HMCL samples were done on U133 Plus 2.0 (Affymetrix, Santa Clara, CA) as previously described (Carrasco et al, 2006). Median normalized gene expression profiles were analyzed using GeneSpring (Agilent).
High-resolution aCGH was performed on 62 patient and 46 HMCL samples with the Human Genome 44B microarray (Agilent Technologies) following the protocol suggested by the manufacturer. Briefly, 800 ng of test (MM CD138+ cells or HMCL) and normal female reference (Promega) were directly labeled with either Cy5 or Cy3 using the BioPrime Array CGH Genomic Labeling Module (Invitrogen) and purified using Vivaspin 500 spin columns (Sartorius). The hybridization reactions containing equal amounts of test and reference DNA were hybridized to the microarray at 65°C for 40 h in a rotation oven at 20 rpm. The slides were washed and then scanned with the Agilent G2505B DNA microarray scanner. The microarray images were analyzed using Feature Extraction software V8.1 (Agilent) and Log2 transformed ratio data was analyzed with GeneSpring GX V7.3.1 and CGH Analytics V3.4.27 (Agilent)
Sequencing
When possible individual coding exons were amplified from 10 ng of CD138 purified plasma cell DNA in 25 ul reactions using Platinum Taq DNA Polymerase (Invitrogen, Carlsbad CA) and M13 tagged exon specific primes (Table S4). Alternatively, overlapping segments of the entire open reading frame were amplified from RNA converted to cDNA using Superscript II and oligo dT17. Sequencing reactions were carried out using Big Dye V3.1 and capillary electrophoresis was performed on an ABI 3730 at the Mayo Clinic or TGEN sequencing core facilities. DNA sequences were analyzed using Sequencher V4.5.
FISH
The cIg-FISH method was performed as previously described(Fonseca et al., 2003a). Briefly, probes specific to each region of interest were selected using the UCSC genome browser and the specific positions were confirmed by manual mapping of the available end sequences to data available from HGB 36.2. The selected BAC clones were purchased from Invitrogen and the Fosmid clones were ordered from the BACPAC Resources Center. The specificity of each probe was confirmed by hybridization to normal metaphase preparations to confirm chromosome and band specificity, and gene specific PCR was used to confirm the presence of DNA sequence corresponding to the specific region of interest. A list of the various FISH probes is provided in Table S5 and a map of the different regions is provided in Figure S11.
Immunoblotting, Immunofluorescence, and Immunohistochemistry
Total cellular extracts were prepared from freshly isolated HMCLs using 10x Cell Lysis Buffer (Cell Signaling Technologies) supplemented with 1mM PMSF. Protein samples were resolved on 7.5% SDS-PAGE gels and transferred to PVDF membranes. The individual blots were blocked with 5% non-fat milk for 1 hr and stained overnight with specific antibodies; TRAF3 H-122 (Santa Cruz), NIK (Cell Signaling), cIAP1 (human) and cIAP2 (human)(R&D Systems), NFKB2 (rabbit anti-p100/p52 N-terminal polyclonal, kindly provided by Warner Greene, UCSF). Protein bands of interest were imaged with HRP-conjugated secondary antibodies.
The localization of NFKB2 in HMCL and selected patient core biopsies was determined with an NFKB2 monoclonal antibody (18D10, Cell Signaling Technology). Fresh cytospins of HMCLs were fixed in 4% paraformaldeyde, permeabilized with 0.1% Triton X-100, and stained with a 1:50 dilution. Confocal slices of 2 μm were collected on a Zeiss LSM510 microscope using a 63x objective (Zeiss, Plan-Apochromat 63x/1.4 oil). Sections from available core biopsies were stained with a 1:50 dilution.
Generation of helper-dependent adenoviral vectors
We created a helper-dependent adenoviral (HDAd) vector precursor plasmid system capable of the simultaneous expression of EGFP and a gene-of-interest from two different promoters near the left and right ITRs, respectively. The original adenoviral plasmids, which have been described previously(Shi et al., 2006), were kindly provided by Mary Hitt (University of Alberta). First, we created the HDAd precursor plasmid, pSC27B, by replacing the beta-galactosidase expression cassette near the left ITR of pSC9B with a murine cytomegalovirus (mCMV) driven EGFP expression cassette. Second, we created the pSC11MCPA shuttle plasmid by introducing a mCMV-MCS-SV40 poly-A cassette in between the I-SceI and I-CeuI restriction sites of pSC11. Thus, allowing a gene-of-interest expression cassette from pSC11MCPA to be transferred to a region near the right ITR of pSC27B.
The full-length TRAF3 cDNA clone (Image Consortium clone 30915387) was purchased from Open Biosystems. The ORF was excised from the pCR4.0-TOPO vector with EcoRI and cloned into the MCS of pSC11MCPA. The expression cassette was excised from pSC11MCPA-TRAF3 with I-CeuI and I-SceI and transferred to pSC27B to create pSC27B-TRAF3. Helper-dependent adenoviral particles were prepared by repeated cycles of amplification in 293Cre4 cells (kindly provided by Frank Graham, McMaster University) with the adeno helper virsus AdRP2050 (Robin Parks, Ottawa Health Research Institute), which contains a capsid modified by insertion of RGD into the H1 loop of the fiber. Virus particles were purified with a 1 hr step gradient and overnight continuous CsCl gradients as described previously (Parks et al., 1996). Virus particles were quantified as described previously (Ng et al., 2002). Healthy HMCLs were infected with 150 viral particles per cell in 60mm Petri plates with 2.5×106 cells in 5 ml of antibiotic free media. The percentage of infected cells was monitored by FACS analysis of the co-expressed GFP marker.
Functional Analysis of TRAF3wt reintroduction
Infected HMCL were counted 24 hours post-infection and seeded at specific densities in triplicate in 96 well plates, for MTT assay, and 6 well dishes; for protein assays, apoptosis, and cell cycle analysis. For protein assays TRAF3wt and control infected cells were isolated 48 hours post infection. For cell cycle analysis cells were stained with propidium iodide and the FACS results were modeled with ModFit LT and chi-square value below 5. The number of apoptotic cells was determined by Annexin-V-Alexa 647 (Invitrogen) staining.
Electronic availability of the Data
A complete list of the patient samples and various tests are provided in Supplementary Table S6. The publicly available datasets used in this paper are available in gene expression omnibus (GEO) or institutional websites as follows, Carrasco et al. GEP (GSE4452) and aCGH (http://genomic.dfci.harvard.edu/array_cgh_db.htm). The GEP data from our patient dataset is available in the GEO database under the accession number GSE6477. The complete dataset is freely available from the Multiple Myeloma Research Consortium Genomics Portal (http://www.broad.mit.edu/mmgp).
Supplementary Material
Acknowledgments
We are indebted to Warner C. Green and Robin J. Parks for so kindly providing the anti-p100/p52 antibody and AdRP2050 helper adenovirus. We would also like to thank the TGen Sequencing Core facility for their assistance with the DNA sequencing. We would also like to thank Paige Anderson, Aprill Watanabe and Sandra Montgomery for their invaluable assistance. This work was supported by grants R01-CA83724-01 (RF), SPORE P50-CA100707-01 (RF and PLB) and P01-CA62242 (RF) from the National Cancer Institute, and the Donaldson Charitable Trust Fund (RF). RF is a Clinical Investigator of the Damon Runyon Cancer Research Fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Annunziata CM, Davis R, Gabrea A, Kuehl M, Staudt LM. NF-kappaB-inducing kinase activates NF-kappaB signaling in multiple myeloma. Proc Amer Assoc Cancer Res. 2006;47 [Abstract #LB-124] [Google Scholar]
- Barrett MT, Scheffer A, Ben-Dor A, Sampas N, Lipson D, Kincaid R, Tsang P, Curry B, Baird K, Meltzer PS, et al. Comparative genomic hybridization using oligonucleotide microarrays and total genomic DNA. Proc Natl Acad Sci U S A. 2004;101:17765–17770. doi: 10.1073/pnas.0407979101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsagel PL, Chesi M, Nardini E, Brents LA, Kirby SL, Kuehl WM. Promiscuous translocations into immunoglobulin heavy chain switch regions in multiple myeloma. Proc Natl Acad Sci U S A. 1996;93:13931–13936. doi: 10.1073/pnas.93.24.13931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsagel PL, Kuehl WM, Zhan F, Sawyer J, Barlogie B, Shaughnessy J., Jr Cyclin D dysregulation: an early and unifying pathogenic event in multiple myeloma. Blood. 2005;106:296–303. doi: 10.1182/blood-2005-01-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caamano JH, Rizzo CA, Durham SK, Barton DS, Raventos-Suarez C, Snapper CM, Bravo R. Nuclear factor (NF)-kappa B2 (p100/p52) is required for normal splenic microarchitecture and B cell-mediated immune responses. J Exp Med. 1998;187:185–196. doi: 10.1084/jem.187.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasco DR, Tonon G, Huang Y, Zhang Y, Sinha R, Feng B, Stewart JP, Zhan F, Khatry D, Protopopova M, et al. High-resolution genomic profiles define distinct clinico-pathogenetic subgroups of multiple myeloma patients. Cancer Cell. 2006;9:313–325. doi: 10.1016/j.ccr.2006.03.019. [DOI] [PubMed] [Google Scholar]
- Ciana P, Neri A, Cappellini C, Cavallo F, Pomati M, Chang CC, Maiolo AT, Lombardi L. Constitutive expression of lymphoma-associated NFKB-2/Lyt-10 proteins is tumorigenic in murine fibroblasts. Oncogene. 1997;14:1805–1810. doi: 10.1038/sj.onc.1201015. [DOI] [PubMed] [Google Scholar]
- Courtois G, Gilmore TD. Mutations in the NF-kappaB signaling pathway: implications for human disease. Oncogene. 2006;25:6831–6843. doi: 10.1038/sj.onc.1209939. [DOI] [PubMed] [Google Scholar]
- Dejardin E. The alternative NF-kappaB pathway from biochemistry to biology: pitfalls and promises for future drug development. Biochem Pharmacol. 2006;72:1161–1179. doi: 10.1016/j.bcp.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Fonseca R, Blood E, Rue M, Harrington D, Oken MM, Kyle RA, Dewald GW, Van Ness B, Van Wier SA, Henderson KJ, et al. Clinical and biologic implications of recurrent genomic aberrations in myeloma. Blood. 2003a;101:4569–4575. doi: 10.1182/blood-2002-10-3017. [DOI] [PubMed] [Google Scholar]
- Fonseca R, Debes-Marun CS, Picken EB, Dewald GW, Bryant SC, Winkler JM, Blood E, Oken MM, Santana-Davila R, Gonzalez-Paz N, et al. The recurrent IgH translocations are highly associated with nonhyperdiploid variant multiple myeloma. Blood. 2003b;102:2562–2567. doi: 10.1182/blood-2003-02-0493. [DOI] [PubMed] [Google Scholar]
- Fotin-Mleczek M, Henkler F, Hausser A, Glauner H, Samel D, Graness A, Scheurich P, Mauri D, Wajant H. Tumor necrosis factor receptor-associated factor (TRAF) 1 regulates CD40-induced TRAF2-mediated NF-kappaB activation. J Biol Chem. 2004;279:677–685. doi: 10.1074/jbc.M310969200. [DOI] [PubMed] [Google Scholar]
- Fracchiolla NS, Lombardi L, Salina M, Migliazza A, Baldini L, Berti E, Cro L, Polli E, Maiolo AT, Neri A. Structural alterations of the NF-kappa B transcription factor lyt-10 in lymphoid malignancies. Oncogene. 1993;8:2839–2845. [PubMed] [Google Scholar]
- Franzoso G, Carlson L, Poljak L, Shores EW, Epstein S, Leonardi A, Grinberg A, Tran T, Scharton-Kersten T, Anver M, et al. Mice deficient in nuclear factor (NF)-kappa B/p52 present with defects in humoral responses, germinal center reactions, and splenic microarchitecture. J Exp Med. 1998;187:147–159. doi: 10.1084/jem.187.2.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grech AP, Amesbury M, Chan T, Gardam S, Basten A, Brink R. TRAF2 differentially regulates the canonical and noncanonical pathways of NF-kappaB activation in mature B cells. Immunity. 2004;21:629–642. doi: 10.1016/j.immuni.2004.09.011. [DOI] [PubMed] [Google Scholar]
- He JQ, Saha SK, Kang JR, Zarnegar B, Cheng G. Specificity of TRAF3 in its negative regulation of the noncanonical NF-kappa B pathway. J Biol Chem. 2006a doi: 10.1074/jbc.M610271200. [DOI] [PubMed] [Google Scholar]
- He JQ, Zarnegar B, Oganesyan G, Saha SK, Yamazaki S, Doyle SE, Dempsey PW, Cheng G. Rescue of TRAF3-null mice by p100 NF-kappa B deficiency. J Exp Med. 2006b;203:2413–2418. doi: 10.1084/jem.20061166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann A, Baltimore D. Circuitry of nuclear factor kappaB signaling. Immunol Rev. 2006;210:171–186. doi: 10.1111/j.0105-2896.2006.00375.x. [DOI] [PubMed] [Google Scholar]
- Ishikawa H, Carrasco D, Claudio E, Ryseck RP, Bravo R. Gastric hyperplasia and increased proliferative responses of lymphocytes in mice lacking the COOH-terminal ankyrin domain of NF-kappaB2. J Exp Med. 1997;186:999–1014. doi: 10.1084/jem.186.7.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- Khaja R, Zhang J, Macdonald JR, He Y, Joseph-George AM, Wei J, Rafiq MA, Qian C, Shago M, Pantano L, et al. Genome assembly comparison identifies structural variants in the human genome. Nat Genet. 2006;38:1413–1418. doi: 10.1038/ng1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuai J, Nickbarg E, Wooters J, Qiu Y, Wang J, Lin LL. Endogenous association of TRAF2, TRAF3, cIAP1, and Smac with lymphotoxin beta receptor reveals a novel mechanism of apoptosis. J Biol Chem. 2003;278:14363–14369. doi: 10.1074/jbc.M208672200. [DOI] [PubMed] [Google Scholar]
- Kuehl WM, Bergsagel PL. Multiple myeloma: evolving genetic events and host interactions. Nat Rev Cancer. 2002;2:175–187. doi: 10.1038/nrc746. [DOI] [PubMed] [Google Scholar]
- Liao G, Zhang M, Harhaj EW, Sun SC. Regulation of the NF-kappaB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. J Biol Chem. 2004;279:26243–26250. doi: 10.1074/jbc.M403286200. [DOI] [PubMed] [Google Scholar]
- Mulligan G, Mitsiades C, Bryant B, Zhan F, Chng WJ, Roels S, Koenig E, Fergus A, Huang Y, Richardson P, et al. Blood. 2006. Gene expression profiling and correlation with outcome in clinical trials of the proteasome inhibitor bortezomib. [DOI] [PubMed] [Google Scholar]
- Neri A, Chang CC, Lombardi L, Salina M, Corradini P, Maiolo AT, Chaganti RS, Dalla-Favera R. B cell lymphoma-associated chromosomal translocation involves candidate oncogene lyt-10, homologous to NF-kappa B p50. Cell. 1991;67:1075–1087. doi: 10.1016/0092-8674(91)90285-7. [DOI] [PubMed] [Google Scholar]
- Ng P, Parks RJ, Graham FL. Preparation of helper-dependent adenoviral vectors. Methods Mol Med. 2002;69:371–388. doi: 10.1385/1-59259-141-8:371. [DOI] [PubMed] [Google Scholar]
- Olshen AB, Venkatraman ES, Lucito R, Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5:557–572. doi: 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
- Parks RJ, Chen L, Anton M, Sankar U, Rudnicki MA, Graham FL. A helper-dependent adenovirus vector system: removal of helper virus by Cre-mediated excision of the viral packaging signal. Proc Natl Acad Sci U S A. 1996;93:13565–13570. doi: 10.1073/pnas.93.24.13565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qing G, Qu Z, Xiao G. Stabilization of basally translated NF-kappaB-inducing kinase (NIK) protein functions as a molecular switch of processing of NF-kappaB2 p100. J Biol Chem. 2005;280:40578–40582. doi: 10.1074/jbc.M508776200. [DOI] [PubMed] [Google Scholar]
- Rajkumar SV, Richardson PG, Hideshima T, Anderson KC. Proteasome inhibition as a novel therapeutic target in human cancer. J Clin Oncol. 2005;23:630–639. doi: 10.1200/JCO.2005.11.030. [DOI] [PubMed] [Google Scholar]
- Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson PG, Barlogie B, Berenson J, Singhal S, Jagannath S, Irwin D, Rajkumar SV, Srkalovic G, Alsina M, Alexanian R, et al. A Phase 2 Study of Bortezomib in Relapsed, Refractory Myeloma. N Engl J Med. 2003;348:2609–2617. doi: 10.1056/NEJMoa030288. [DOI] [PubMed] [Google Scholar]
- Sen R. Control of B lymphocyte apoptosis by the transcription factor NF-kappaB. Immunity. 2006;25:871–883. doi: 10.1016/j.immuni.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Shi CX, Graham FL, Hitt MM. A convenient plasmid system for construction of helper-dependent adenoviral vectors and its application for analysis of the breast-cancer-specific mammaglobin promoter. J Gene Med. 2006;8:442–451. doi: 10.1002/jgm.867. [DOI] [PubMed] [Google Scholar]
- Shinkura R, Kitada K, Matsuda F, Tashiro K, Ikuta K, Suzuki M, Kogishi K, Serikawa T, Honjo T. Alymphoplasia is caused by a point mutation in the mouse gene encoding Nf-kappa b-inducing kinase. Nat Genet. 1999;22:74–77. doi: 10.1038/8780. [DOI] [PubMed] [Google Scholar]
- Vischioni B, Giaccone G, Span SW, Kruyt FA, Rodriguez JA. Nuclear shuttling and TRAF2-mediated retention in the cytoplasm regulate the subcellular localization of cIAP1 and cIAP2. Exp Cell Res. 2004;298:535–548. doi: 10.1016/j.yexcr.2004.04.040. [DOI] [PubMed] [Google Scholar]
- Zapata JM, Krajewska M, Krajewski S, Kitada S, Welsh K, Monks A, McCloskey N, Gordon J, Kipps TJ, Gascoyne RD, et al. TNFR-associated factor family protein expression in normal tissues and lymphoid malignancies. J Immunol. 2000;165:5084–5096. doi: 10.4049/jimmunol.165.9.5084. [DOI] [PubMed] [Google Scholar]
- Zender L, Spector MS, Xue W, Flemming P, Cordon-Cardo C, Silke J, Fan ST, Luk JM, Wigler M, Hannon GJ, et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell. 2006;125:1253–1267. doi: 10.1016/j.cell.2006.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.