

Abstract
The surface transmembrane glycoprotein is responsible for mediating virion attachment to cell and subsequent virus-cell membrane fusion. However, the molecular mechanisms for the viral entry of coronaviruses remain poorly understood. The crystal structure of the fusion core of mouse hepatitis virus S protein, which represents the first fusion core structure of any coronavirus, reveals a central hydrophobic coiled coil trimer surrounded by three helices in an oblique, antiparallel manner. This structure shares significant similarity with both the low pH-induced conformation of influenza hemagglutinin and fusion core of HIV gp41, indicating that the structure represents a fusion-active state formed after several conformational changes. Our results also indicate that the mechanisms for the viral fusion of coronaviruses are similar to those of influenza virus and HIV. The coiled coil structure has unique features, which are different from other viral fusion cores. Highly conserved heptad repeat 1 (HR1) and HR2 regions in coronavirus spike proteins indicate a similar three-dimensional structure among these fusion cores and common mechanisms for the viral fusion. We have proposed the binding regions of HR1 and HR2 of other coronaviruses and a structure model of their fusion core based on our mouse hepatitis virus fusion core structure and sequence alignment. Drug discovery strategies aimed at inhibiting viral entry by blocking hairpin formation may be applied to the inhibition of a number of emerging infectious diseases, including severe acute respiratory syndrome.
Coronaviruses are enveloped viruses with single-stranded, positive-sense genomic RNA that is 26–31 kb in length (1, 2). The Coronaviridae exhibit a broad host range, infecting many mammalian and avian species and causing upper respiratory, hepatic, gastrointestinal, and central nervous system diseases. Coronaviruses in humans and fowl primarily cause upper respiratory tract infections, whereas porcine and bovine coronaviruses establish enteric infections that result in severe economic loss (3). The coronaviruses also include mouse hepatitis virus (MHV),1 infectious bronchitis virus, feline infectious peritonitis virus, and the newly emergent human severe acute respiratory syndrome-associated coronavirus (HcoV-SARS) (4).
The surface glycoproteins of enveloped viruses play essential roles in viral entry into cells by mediating virion attachment to cells and the virus-cell membrane fusion, the initial events of the viral infections. The spike (S) protein is the sole viral enveloped glycoprotein responsible for cell entry in coronaviruses. It binds to the cell surface receptor and mediates subsequent fusion of the viral and cellular membranes (5). Under the electron microscope, the spike proteins can be clearly seen as 20-nm-long surface projections on the virion membrane (6).
The spike proteins of coronaviruses share several features with other viral glycoproteins mediating viral entry, including the hemagglutinin (HA) protein of influenza virus, gp160 of human immunodeficiency virus (HIV) and simian immunodeficiency virus (SIV), GP of Ebola virus, and fusion protein of paramyxovirus (7, 8). These glycoproteins are all synthesized as single polypeptide precursors that oligomerize in the endoplasmic reticulum to form trimers. Most of the enveloped proteins with fusion activity contain two noncovalently associated subunits: S1 + S2 in coronaviruses, HA1 + HA2 in influenza viruses, gp120 + gp41 in HIV/SIV, GP1 + GP2 in Ebola virus, and F1 + F2 in paramyxoviruses, all of which are generated by proteolytic cleavage. Nevertheless, some enveloped proteins have no cleavage in their precursors and yet still maintain fusion activity, such as the S proteins of some coronaviruses (9) and GP of Ebola virus (10).
A hydrophobic region in the membrane-anchored subunit of enveloped proteins, termed the fusion peptide in class I fusion proteins, has been shown to insert into the cellular membranes during the fusion process (11, 12). The regions following the fusion peptide have a 4-3 heptad repeat (HR) of hydrophobic residues, a sequence feature characteristic of coiled coils. The first heptad repeat, termed HR1 (HRA or N peptide), is followed by a short spacer domain and a second heptad repeat, termed HR2 (HRB or C-peptide), followed by another short spacer and the transmembrane (TM) anchor (13). Biochemical and structural analyses of HA2 (14), HIV-1/SIV gp41 (15, 16, 17, 18, 19, 20, 21), Ebola virus GP2 (22, 23), and SV5F1 (24) indicate that these heptad repeat regions form six-helix bundles. The N-terminal heptad repeat forms a central coiled coil, which is surrounded by three HR2 helices in an oblique, antiparallel manner.
Among all enveloped glycoproteins, the membrane fusion mechanism of the HA of influenza viruses has been studied in greater detail (7, 8, 25). HA is proteolytically cleaved to generate a receptor binding subunit (HA1) and an anchored subunit (HA2) containing the fusion peptide. Numerous evidences suggest that the HA of influenza undergoes a conformational change, from a native (nonfusogenic) to fusion-active (fusogenic) state during the viral fusion process. In the native HA, part of the heptad repeat region of HA2 forms a nonhelical loop (26) but converts into a coiled coil when exposed to low pH (14). The later conformation is generally regarded to be a fusogenic state because the low pH also activates influenza membrane fusion. This conformation change is also the basis of the “spring-loaded mechanism” for activation of viral fusion (27).
The spike protein of MHV A59 has been identified as a class I fusion protein and shares common features with other viral fusion proteins (28). However, there are several significant differences between the membrane-anchored subunits of coronavirus spike proteins and HA2, gp41, GP2, and SV5F1. First, the ectodomain is much larger (550 residues versus 120–380 residues) in size in S protein. Second, the HR1 region is predicted to be much larger (more than 100 residues versus 30–60 residues) in size by the learn-coil VMF program (29) and also verified by proteinase K-resistant experiments (28). Third, the putative fusion peptides of all other viral fusion proteins are located at the N terminus of the membrane-proximal subunits, whereas the S protein features an internal fusion peptide.
Highly conserved HR1 and HR2 regions in coronavirus spike proteins suggest that they share similar three-dimensional fusion core structures and a common mechanism for viral fusion. The binding region of the HR1·HR2 complex of other coronaviruses and likely structures of their fusion cores can be proposed based on MHV fusion core structure and sequence alignment. Analogous to HIV C-peptides, HR2 peptides of coronaviruses are likely to act in a dominant-negative manner to inhibit hairpin formation, thereby inhibiting viral entry. Thus, drug discovery strategies aimed at inhibiting viral entry by blocking hairpin formation may also be applied to the inhibition of emerging infectious diseases such as SARS.
Although previous biochemical and electron microscopic analyses have shown that the HR1·HR2 complex in the S protein of MHV forms a fusion core (28), the exact binding region of the HR1·HR2 complex and the detailed structure of the fusion core remain unknown. Here we report the determination of the crystal structure of the fusion core of MHV A59 spike protein to 2.5 Å resolution by x-ray crystallography and discuss the implications of the structure for coronavirus membrane fusion.
EXPERIMENTAL PROCEDURES
Expression, Purification, and Crystallization—The expression, purification, and preliminary crystallographic studies of the MHV 2-Helix protein have been described elsewhere.2 The PCR-directed gene was inserted into pET22b (Novagen) vector, and the selenomethionine MHV 2-Helix derivative was expressed in M9 medium containing 60 mg liter-1 selenomethionine in Escherichia coli strain BL21 (DE3). The product was purified by nickel-nitrilotriacetic acid affinity chromatography followed by gel filtration chromatography. The purified MHV 2-Helix derivative was dialyzed against 10 mm Tris, pH 8.0, 10 mm NaCl and concentrated to 8 mg ml-1. Crystals with good diffracting quality could be obtained in 0.1 m MES, pH 6.5, 10% PEG 4000 (v/v), 8% dimethyl sulfoxide (v/v), 5 mm hexaminecobalt trichloride after 3 days. The expression, purification, and crystallization of nMHV is the same as for MHV 2-Helix. Crystals with good diffracting quality could be obtained in 0.1 m MES, pH 6.5, 13% PEG 4000 (v/v), 5% dimethyl sulfoxide (v/v).
Data Collection and Processing—The MHV 2-Helix crystal was mounted on nylon loops and flash-frozen in an Oxford Cryosystems cold nitrogen gas stream at 100 K. Multiple wavelength anomalous dispersion data were collected by a rotation method using a MarCCD detector with synchrotron radiation on beamline 3W1A of the Beijing Synchrotron Radiation Facility. Data were collected from a single selenomethionyl derivative crystal at peak (0.9799 Å), inflection (0.9801 Å), and remote (0.9000 Å) wavelengths to 2.5 Å. Data collection from the nMHV 2-Helix crystal was performed in-house on a Rigaku RU2000 rotating anode x-ray generator operated at 48 kV and 98 mA (Cu Kα; λ = 1.5418 Å) with a MAR 345 image plate detector. The crystal was mounted on nylon loops and flash-frozen at 100 K using an Oxford Cryosystems cold nitrogen gas stream. Data were indexed and scaled using DENZO and SCALEPACK programs (30). Data collection statistics are shown in Table I .
Table I.
Data collection and model refinement statistics
| Data set statistics | ||||
|---|---|---|---|---|
| MHV 2-Helix |
nMHV 2-Helix native | |||
| Peak | Edge | Remote | ||
| Space group | R3 | R3 | R3 | R3 |
| Unit cell parameters (Å) | 48.4 48.4 200.0 | 48.47 48.47 200.1 | 48.3 48.3 199.4 | 51.6 51.6 198.2 |
| Wavelength (Å) | 0.9799 | 0.9801 | 0.9000 | 1.5418 |
| Resolution limit (Å) | 2.4 | 2.4 | 2.4 | 2.06 |
| Observed reflections | 49,717 | 50,167 | 49,352 | 83,487 |
| Unique reflections | 6,842 | 6,867 | 6,804 | 12,068 |
| Completeness (%) | 100(100)a | 100(100) | 100(100) | 98.6(100) |
| 〈I/σ(I)〉 | 14.3(4.1) | 14.6(4.5) | 13.7(3.8) | 28.7(4.3) |
| Rmergeb (%) | 9.4(34.0) | 8.4(31.0) | 9.3(36.1) | 9.4(29.9) |
| Final refinement statistics | ||
|---|---|---|
| MHV 2-Helix | nMHV 2-Helix | |
| Rworkc (%) | 22.4 | 26.2 |
| Rfreed (%) | 29.1 | 29.8 |
| Resolution range (Å) | 35–2.5 | 50–2.06 |
| Total reflections used | 5,833 | 11,689 |
| No. of reflections in working set | 5,501 | 11,092 |
| No. of reflections in test set | 332 | 597 |
| r.m.s.d.e bonds(Å) | 0.012 | 0.012 |
| r.m.s.d. angles(°) | 1.8 | 1.4 |
Numbers in parentheses correspond to the highest resolution shell
Rmerge = ΣhΣlIih – 〈Ih〉 /ΣhΣI 〈Ih〉, where 〈Ih〉 is the mean of the observations Iih of reflection h
Rwork = Σ(∥Fobs – Fcalc∥)/Σ Fobs
Rfree is the R-factor for a subset (5%) of reflections that was selected prior refinement calculations and not included in the refinement
r.m.s.d., root mean square deviation from ideal geometry
Phase Determination and Model Refinement—For MHV 2-Helix structural determination, initial multiple wavelength anomalous dispersion phasing steps were performed using SOLVE (31) and followed by density modification by RESOLVE (32). The program O (33) was used for viewing electron density maps and manual building. The initial structure was subsequently refined to a final R-value of 22.4% and free R-value of 29.1%. The quality of the structure was verified by PRO-CHECK (34). None of the main chain torsion angles is located in disallowed regions of the Ramachandran plot. The statistics for the structure determination and refinement are summarized in Table I. The figures were generated with the programs GRASP (35), SPDBView (36), and MOLSCRIPT (37).
RESULTS AND DISCUSSION
Structure Determination—Two peptides (HR1 and HR2, Fig. 1, A and B ) encompassing the N-terminal and C-terminal heptad repeats of the MHV spike protein assemble into a stable trimer of heterodimers (28). The HR1 and HR2 regions of the MHV S protein consist of residues 968–1027 and residues 1216–1254, respectively (Fig. 1, A and B). The fusion core of the MHV spike protein was prepared as a single chain by linking the HR1 and HR2 domains via an eight-amino acid linker (GGSGGSGG, single amino acid abbreviation used here). The constructs and the encoded proteins were also called MHV 2-Helix (Fig. 1A). The preparation and characterization of the 2-Helix proteins will be reported elsewhere.2 The MHV 2-Helix forms crystals that have unit cell parameters a = b = 48.3 Å, c = 199.6Å, α = β = 90°, γ = 120° and belong to the space group R3. The crystals contain two MHV 2-Helix molecules/asymmetric unit, and the diffraction pattern extends to 2.5 Å resolution.2
Fig. 1.
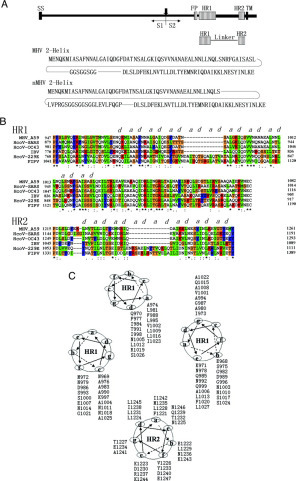
Structure determination of the MHV spike protein fusion core trimer.A, schematic representation of coronavirus MHV A59 spike protein and the MHV 2-Helix and nMHV 2-Helix constructs. S1 and S2 are formed after proteolytic cleavage (vertical arrow) and noncovalently linked. The enveloped protein has an N-terminal signal sequence (SS) and a TM domain adjacent to the C terminus. S2 contains two HR regions (hatched bars), termed HR1 and HR2 as indicated. FP (hatched bars) is a putative fusion peptide followed by HR1 region. For the MHV 2-Helix, two HR regions were linked to a single polypeptide with an 8-residue linker (GGSGGSGG). For the nMHV 2-Helix, HR2 and a shortened HR1 were linked with a 22-amino acid linker (LVPRGSGGSGGSGGLEVLFQGP). B, sequence alignment of coronavirus spike protein HR1 and HR2 regions. Letters above the sequence indicate the predicted hydrophobic HR a and d residues, which are highly conserved. C, helical wheel representation of HR1 and HR2. Three HR1 helices and one HR2 helix are represented as helical wheel projections. The view is from the top of the structure. The three central HR1 helices form a central hydrophobic core with the interaction of residues in the a and d positions. The three HR2 helices pack against these hydrophobic surface grooves through interactions with residues in the a and d positions in HR2 and e and g positions in HR1. These residues, mediating the interactions between HR1 and HR2, are always hydrophobic and conserved (see B).
The crystal structure of the MHV 2-Helix was solved by multiple wavelength anomalous dispersion from a single selenomethionyl derivative crystal. Four selenium sites could be located in one asymmetric unit from Patterson maps calculated using the program CNS 1.0 (38). The experimental electron density map was easily interpretable in the helical regions. The model was improved further by cycles of manual building and refinement using the programs O (33) and CNS (38). The structure was subsequently refined to a final resolution of 2.5 Å with an R-value of 22.4% and free R-value of 29.1%. The final model statistics are summarized in Table I.
Description of the Structure—In the three-dimensional structure of the MHV 2-Helix, the fusion core has a rod-shaped structure ∼80 Å in length and a maximum diameter of 28 Å. The complex is a six-helix bundle comprising a trimer of MHV 2-Helix. The center of this bundle consists of a parallel trimeric coiled coil of three HR1 helices that were packed by three antiparallel HR2 helices (Fig. 2, A and B ). The N terminus of HR1 and the C terminus of HR2 are located at the same end of the six-helix bundles, placing the fusion peptide and TM domains close together. A region of about 190 amino acids would be located at the other end of the six-helix bundle between HR1 and HR2. The linker and several terminal residues were disordered in both molecules. In one asymmetric unit, one molecule includes residues 970–1023 in HR1 and 1216–1252 in HR2, and the other molecule includes residues 969–1022 in HR1 and 1216–1254 in HR2. The two trimers are created by the same 3-fold axis of the crystallographic unit cells and are both parallel with the 3-fold axis of the crystallographic unit cells, and there is about 30 degrees rotation in the orientation parallel with the 3-fold axis between the two trimers. The root mean square deviation of the two molecules in one asymmetric unit is 0.36 Å. There is only one weak hydrogen bond between the two parallel trimers, from OH (Tyr1233) to O (Glu1254) with a distance of 3.34 Å.
Fig. 2.
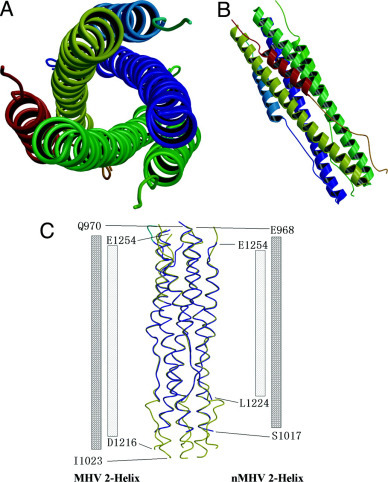
Overall views of the fusion core structure and superposition of nMHV (new construct for MHV fusion core) and MHV fusion core.A, top view of the MHV fusion core structure showing the 3-fold axis of the trimer. B, side view of the MHV fusion core structure showing the six-helix bundle. C, side view showing the superposition of nMHV fusion core (colored in blue) and MHV fusion core (colored in yellow). The columns at both sides of the map represent two HR1 and HR2 regions of nMHV and MHV fusion cores. The number at the end of these columns represents the end residues in the two structures.
Residues 969–1022 of HR1 fold into a 15-turn α-helix stretching the entire length of the coiled coil. As in other naturally occurring coiled coils of the fusion core, the residues in the a and d positions of HR1 are predominantly hydrophobic (Fig. 1B). A sequence alignment of MHV with other representative coronavirus spike proteins shows that the residues at these two heptad repeat positions are highly conserved (Fig. 1B).
Residues 1232–1247 of HR2 form a 5-turn amphipathic α-helix, whereas residues 1216–1231 and 1248–1254 form two extended chains at the N and C terminus of HR2, respectively. Each HR2 fits into the long grooves formed by the interface of the three HR1 helices, and no interaction is observed between individual HR2 helices (Fig. 2, A and B). The C terminus of HR2 ends with Glu1254, which is aligned with Gln970 of HR1; Gln970 is also the N terminus of the HR1 domain. The N terminus of HR2 starts with Asp1216, which is aligned with Ile1023 of HR1 (Fig. 2C).
Linker between HR1 and HR2 and nMHV 2-Helix Structure—To verify whether the linker (GGSGGSGG) between HR1 and HR2 affects the natural structure of the MHV 2-Helix, we made a new construct (termed nMHV 2-Helix) that includes a shorter HR1 and longer linker. In the nMHV 2-Helix construct, the HR1 consists of residues 968–1017, the HR2 consists of residues 1216–1254, and the new fusion core was prepared as a single chain by linking the HR1 and HR2 domains via a 22-amino acid linker (LVPRGSGGSGGSGGLEVLFQGP), which is flexible and long enough to allow the HR1 and HR2 to form a natural interaction (Fig. 1A). The nMHV 2-Helix forms crystals in space group R3 with lattice dimensions of a = b = 51.6 Å, c = 198.2 Å, α = β = 90°, γ = 120°. The crystals contain two 2-Helix molecules/asymmetric unit and diffract x-rays to a resolution of at least 2.1 Å.
The crystal structure of nMHV 2-Helix was determined by molecular replacement with the MHV 2-Helix structure as a search model. Rotation and translation function searches were performed in CNS (38). The model was improved further by cycles of manual building and refinement using the programs O (33) and CNS (38). The final R-value and free R-value for the refinement are 26.2 and 29.8%, respectively. The final model statistics are listed in Table I.
The nMHV 2-Helix structure is largely similar to the MHV 2-Helix structure, with the exception of several residues at the N terminus of HR2 (Fig. 2C). The overall root mean square deviation between the two structures is 0.48 Å, which is calculated using the CCP4 program LSQKAB. The nMHV 2-Helix structure also contains two molecules/asymmetric unit. One molecule includes residues 968–1017 in HR1 and 1224–1254 in HR2, whereas the other molecule includes residues 970–1017 in HR1 and 1229–1254 in HR2. The linker is also disordered and cannot be traced in the structure. The N terminus of HR2 cannot be seen in the structure because the C terminus of HR1, which is important for binding the N terminus of HR2, has been discarded in the new construct. The linker is long and flexible enough, so we can conclude that both structures surely represent the natural structure of the complex of HR1 and HR2 because the choice of linker does not affect the real interaction between the two heptad repeat regions. We will focus our following structural analysis on MHV 2-Helix structure.
Interactions between HR1 and HR2—Three HR2 helices pack obliquely against the outside of the HR1 coiled coil trimer in an antiparallel orientation. The HR2 helices interact with HR1 mainly through hydrophobic residues in three grooves on the surface of the central coiled coil trimer (see Fig. 4A ). Sequence comparison between MHV and other coronavirus spike proteins shows that residues contributing to the HR1/HR2 interaction (e and g positions in HR1, a and d positions in HR2) are highly conserved (Fig. 1B).
Fig. 4.
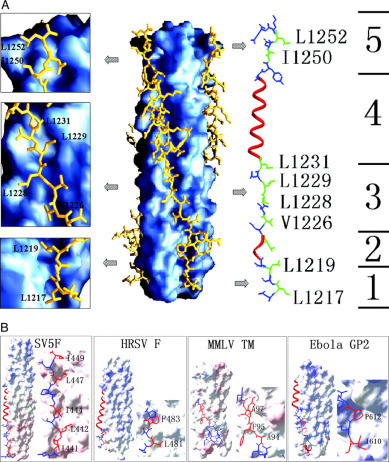
O-X-O motifs in HR2 regions of MHV and the comparison with those of other fusion proteins.A. Left and center, surface map showing the hydrophobic grooves on the surface of three central HR1 helices. Three HR2 helices pack against the hydrophobic groove in an antiparallel manner. The helical regions in HR2 extended regions could be observed clearly. The helical region of HR2 just packs against the deep groove, and the extended region packs against the shallow groove. Right, detailed structure of O-X-O motifs in MHV HR2 region. One HR2 helix is divided into five parts based on its secondary structure. The helical regions (parts 2 and 4) HR2 are colored in red, and extended regions (parts 1, 3, and 5) are colored in blue. The essential residues of the three extended regions and O-X-O motifs in these regions are shown; residues colored in green represent the hydrophobic residues in O-X-O motifs. The three panels on the left show the enlarged images of parts 1, 3, and 5. The hydrophobic residues in these motifs are all packed against the hydrophobic grooves on the surface of three HR1 helices. B, detailed structures of O-X-O motifs in other fusion proteins including SV5F, HRSV F, MMLV Env-TM, and Ebola GP2. They all contain similar motifs in HR2 regions. The regions in which O-X-O motifs are located form extended regions but not α-helices, in a way similar to the MHV 2-Helix.
This pattern of sequence conservation can also be shown by a helical wheel representation of three HR1 helices and one HR2 helix (Fig. 1C). In this diagram, residues in the a and d positions of HR2 pack against residues in the e and g positions of HR1, mainly through hydrophobic interactions. Sequence comparison between MHV and HcoV-OC43 spike proteins shows that no nonconservative changes exist at the e and g positions of HR1, and only three such changes (L to I, L to F and I to L) occur in HR2 at the a and d positions. In contrast, 7 of 26 nonconservative changes occur at the outside f, b, and c positions in HR1, and 5 of 12 nonconservative changes occur at positions other than a and d in the helical region (1232–1248) of HR2 (Fig. 1B).
Comparison with Other Fusion Protein Structures—The structure of the MHV fusion core was compared with the known structures of other viral fusion proteins (Fig. 3A ). Although there are significant differences in the sequences, sizes, and structural properties of these viral fusion proteins, the similarities in their overall structures suggest a common mechanism for membrane fusion. These fusion core proteins are all trimeric coiled coils with the putative fusion peptides located at the N-terminal end of HR1 and the TM domains located at the C-terminal end of HR2 (7). The three HR2 polypeptides that form the outer layer of the central core vary in conformation among these structures, but they always form α-helices and always pack antiparallel to the interior coiled coil. In this pattern, which has been proposed to be a fusogenic conformation, all of these fusion proteins would have their fusion peptides and TM anchors aligned at the same end of the coiled coil.
Fig. 3.
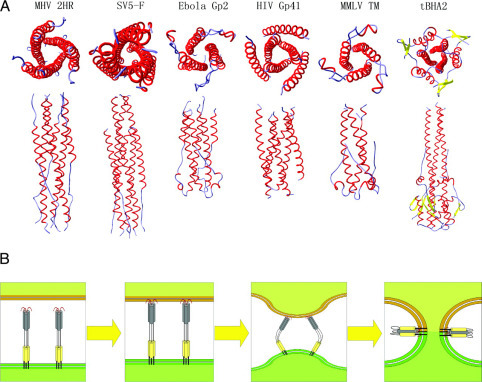
Viral fusion proteins and models for membrane fusion.A, comparison of MHV fusion core with other viral fusion protein structures. The proteins under comparison include SV5F, Ebola GP2, HIV gp41, MMLV Env-TM, and low pH-induced influenza virus HA, tBHA2. Top and side views are shown for the six fusion core structures. B, model for coronavirus-mediated membrane fusion. The first state is the native conformation of coronavirus spike protein on the surface of viral membrane. It has been reported that the spike protein is trimeric in this conformation and about 200 Å in length (6), but the exact structure of the full-length protein remains unknown. The second state is the prehairpin state of the S2 subunit. After several conformational changes, the fusion peptide inserts into the cellular membrane with the aid of other regions of S protein and possibly including the receptor. Although the internal fusion peptide is not exposed at the N-terminal of S2, it could insert into part of the target membrane by means of some hydrophobic residues. This insertion would be stable enough to drive the membrane motion with the conformational changes of HR1 region, which is adjacent to the fusion peptide. The third state is conformational change and juxtaposition of the target and viral membranes. With the help of other regions of S protein, the HR1 and HR2 regions move together and facilitate juxtaposition of the cellular and viral membrane. The last state is the postfusion conformation. The coiled coil will reorient with its long axis parallel to the membrane surface. The fused cellular and viral membranes make it possible for subsequent viral infections.
There are three major differences between the MHV and other viral fusion core structures and their relevant regions, indicating unique features of fusion core structure in coronavirus S proteins. First, the conformation of HR2 in the fusion core structure is different from those of all other fusion core HR2 regions (Fig. 3A). In the MHV fusion core structure, a major 5-turn α-helix and a single-turn α-helix could be observed, and the remaining parts are extended segments. We can divide the HR2 polypeptide into five parts (Fig. 4A, right), parts 1–5. Part 4 is a typical 3-4-3-4-3 pattern and forms a 5-turn α-helix of which the residues at positions a and d pack against residues at the e and g positions of the HR1. Part 2 is also an α-helix that exhibits the 3-4 spacing pattern, although it has only four residues, FEKL. Of these four residues, the residues in the a position (Phe1221) and d position (Leu1224) also pack against hydrophobic grooves formed on the surface of the HR1 core. Parts 1, 3, and 5 should also be α-helical based on prediction by the learn-coil VMF program (29). However, the structure shows they are all extended to form a strand-like conformation. In these three parts, there is an interesting O-X-O motif where O represents hydrophobic residues, and X represents any residue but is generally hydrophilic. Part 1 contains the residues LSL, part 3 contains the residues VTL and LDL, and part 5 contains the residues INL. The two hydrophobic residues in these motifs pack against hydrophobic grooves on the surface of the HR1 core, either facing the central core or aligning with the hydrophobic groove. As a result, the O residues form hydrophobic interactions to stabilize the six-helix bundle, leaving the X residue directed into solvent. This pattern, which we think is a major reason why these three parts do not form α-helices, is also observed in the structure of SV5F (24), HRSV F (39), MMLV TM (40) and Ebola GP2 (41) (Fig. 4B). In these structures, partial regions in HR2 or C-peptide are extended and strand-like rather than α-helical. In the 3-4-3-4-3 pattern in HR2 of these glycoproteins, the presence of an O-X-O motif would result in hydrophobic residues interacting with the hydrophobic grooves on the surface of HR1 core, thus destroying the typical α-helix. In these three-dimensional structures, the distance between the two hydrophobic residues in the O-X-O motifs is about 5 Å, which is also the distance between the two adjacent helices. Thus, the two hydrophobic residues could exactly pack against the grooves of the central coiled coil formed by three HR1 helices. This compatibility of HR2 segments makes the fusion core more stable in solvent because most of the hydrophobic residues in HR2 are packed against the central core, leaving the hydrophilic residues exposed to solvent. This pattern also explains why not all residues in HR2 of fusion cores from many other viral proteins form α-helices and why HR1 structures of these fusion cores are highly conserved, whereas HR2 regions always differ in their three-dimensional conformations (Fig. 3A).
Second, a proteinase K-resistant fusion core of MHV A59 spike protein has been reported by Bosch et al. (28). After digestion by proteinase K, the fusion core comprising residues 958–1040 in HR1 and 1216–1254 in HR2 remains intact. In this fusion core, the HR1 region is about 30 residues longer than the HR1 region of the fusion core we constructed. Although the fusion core structure we determined here is only part of the proteinase K-resistant fusion core, we propose that residues 958–967 and 1023–1040 in HR1 would also form coiled coils in the natural fusion core on the basis of their resistant capacity and other biochemical analyses (28). In the proteinase K-resistant fusion core of MHV spike protein, the central coiled coil HR1 region has about 80 amino acids, which is considerably longer than HR1 segments in other fusion cores such as HIV gp41 and SV5F1. This length is comparable with that of the fusion core in influenza HA, whose mechanisms for the membrane fusion have been studied in extensive detail (25). In addition, the HR1 region of the MHV spike protein is predicted to contain more than 100 amino acids by the learn-coil VMF program (29). This long helical coiled coil might be consistent with the long sequence of S protein, which is more than 1200 residues, to form the central skeleton of spikes on the surface of coronavirus.
Third, although the fusion peptide is not part of the fusion core, it is also very important for investigating viral fusion mechanism. The putative fusion peptide of the MHV A59 spike protein is located at the N-terminal end of the HR1 region, and the cleavage site of the spike protein is about 250 amino acids away from the fusion peptide (28). The cleavage sites of other class I viral fusion proteins are all typically located adjacent to the fusion peptides (13). In the latter pattern, the likely role of the six-helix bundle structure is to facilitate juxtaposition of the viral and cellular membranes by bringing the fusion peptide, which inserts into the cellular membrane, close to the transmembrane segment, which is anchored in the viral membrane (7). In the case of the MHV spike protein, the question of why the HR1 region and fusion peptide are so far away from the cleavage site remains unknown. Nevertheless, some viruses such as coronavirus (9) and Ebola virus (10) do not have cleavage sites in fusion proteins but still retain their fusion activity. This indicates that the location of the fusion peptides, whether exposed at the N terminus of the membrane-anchored polypeptide or not, is not an essential requirement for the viral fusion. We will give a possible mechanism in the further discussion.
Evidence for the Conformational Change—Structural studies of the influenza virus HA and HIV gp41 have established a paradigm for understanding the mechanisms of viral and cellular membrane fusion (7). For coronaviruses, direct evidence for the conformational change in spike protein is lacking, although the crystal structure of the MHV fusion core bears similarity to these fusion-active state molecules. The structure of the MHV 2-Helix could correspond to the fusion core of MHV spike protein in either the fusogenic or the native form of the envelope glycoprotein, or both. Several considerations provide good evidence that the fusion core in the crystal structure presented here is the final, stable form of the protein, which is a fusion-active state following one or more conformational changes.
First, the fusion core of MHV spike protein is exceedingly stable to both thermal denaturation and proteinase K digestion (28). The six-helix bundle has a melting temperature of about 85 °C and could not be separated in general SDS-PAGE unless boiled at 100 °C with a loading buffer containing a high concentration of SDS (28). These properties indicate that the complex is very stable and could not be dissociated by any biologically relevant interaction. This form of fusion core must be present at the later stage of conformational changes for viral fusion, although it is not known whether the complex maintains this conformation throughout the entire process.
Second, virus-cell entry inhibition and cell-cell fusion inhibition experiments (28) also provide strong evidence that the fusion core is formed after one or more conformational changes. HR2 of the MHV 2-Helix could block viral entry and cell-cell fusion in a concentration-dependent manner and appears to be a potent inhibitor (28). In HIV gp41, the C-peptide and its derivatives have been shown to act as dominant-negative inhibitors by binding to the endogenous N-peptide coiled coil trimer within viral gp41 (19, 42, 43). A reasonable interpretation of the data for the MHV fusion core is that HR2 functions in a dominant-negative manner similar to the C-peptide in HIV gp41 by binding to the transiently exposed coiled coil in the prehairpin intermediate and thus preventing the conformational changes required for viral fusion.
Third, mutations in the MHV spike protein that abolish viral-cell fusion activity often map to the HR1 or HR2 residues, which are expected to stabilize the fusion core structure reported here. These studies show that mutations in some essential positions in HR regions abolish infectivity and membrane fusion (44, 45). The L981K and F977K mutations are particularly noteworthy because cells expressing mutant spike proteins with one of these mutations are almost completely defective in membrane fusion, although the surface expression level of spike proteins remains the same as for the wild type (44). Residue Leu981 is in the a position and Phe977 in the d position of the HR1 peptide, and thus both are essential for the formation of the central hydrophobic coiled coil (Fig. 1C). The L981K and F977K mutations in HR1 region substitute hydrophobic residues with hydrophilic residues, destroying the hydrophobic interaction and the formation of the six-helix bundles and subsequently abolishing the membrane fusion. In contrast, replacement of the same residues with other hydrophobic amino acids (F977L, L981I) does not reduce fusion activity (44). L1224A/L1231A, L1224A/I1238A, L1224A/L1245A, and I1231A/L1245A mutations in HR2 also abolish fusion activity greatly (45). In these double substitution mutants, the Leu and Ile residues are both in d positions in HR2 and are also very important for the formation of the fusion core (Fig. 1C). These double substitution mutants could not maintain a stable coiled coil structure even though Leu or Ile was changed for Ala, a hydrophobic residue. The locations of these particular mutations indicate that the interactions between HR1 and HR2 are critical for membrane fusion.
Lastly, the structural similarities between the MHV 2-Helix complex, the fusion core of low pH-induced conformation of influenza HA2 (14), and the structure of HIV gp41 (19), each of which has been proposed to have fusion-active conformations, indicate that the MHV 2-Helix structure studied here represents the core of the fusogenic conformation of spike protein after conformational change (Fig. 3A). In all three structures, the putative hydrophobic fusion peptides are located close to the N-terminal end of the HR1 region, which forms a central coiled coil. Three strands of HR2 which pack against the coiled coil trimer in an antiparallel manner stabilize this hydrophobic coiled coil. These common features suggest that the MHV spike protein also possesses a conformational change mechanism similar to influenza HA and HIV gp41 (7).
Implications for Models of Membrane Fusion Mechanisms— Although we have no structures of full-length MHV spike protein either in the prefusion or postfusion state, we can propose a model for MHV membrane fusion mechanism based on the fusion core structure studied here and previous analysis of spike proteins. Current models for the class I viral fusion mechanisms suggest that the exposed fusion peptide located at the N terminus of the membrane-anchored subunit may be important for the juxtaposition of two membranes prior to fusion (7). For coronaviruses, the S2 fragment contains a putative internal fusion peptide that is not exposed at the N terminus (44). This pattern of internal fusion peptide in the MHV spike protein is reminiscent of fusion loops in class II viral fusion proteins, which are internally located in the fusion protein (46, 47). Recent structural studies of the dengue virus envelope protein show that the highly conserved internal fusion loop penetrates only ∼6 Å into the hydrocarbon layer of the cellular membrane (46). In this structure, an aromatic anchor formed by Trp101 and Phe108 inserts into the cellular membrane. Studies of a 20-residue influenza virus A fusion peptide with a detergent micelle suggests a kinked α-helix, with the N and C termini embedded in the outer leaflet and the kink on the surface (48).
Both the fusion loop from dengue virus E protein and the fusion peptide from influenza virus indicate that the anchoring into the lipid bilayer may not require complete insertion of the fusion peptide into the membrane. The exposure of the fusion peptide at the N terminus of the fusion protein is also not essential for the viral fusion. In this case, which also applies to several coronavirus spike proteins, the fusion peptides may insert into the membrane via several hydrophobic resides or a kinked hydrophobic loop (Fig. 3B). In our model shown in Fig. 3B, the internal fusion peptide of spike proteins forms a small hydrophobic core, which would insert into the membrane after several conformational changes induced by receptors or pH changes. HR2 and HR1 would then form a coiled coil to facilitate juxtaposition of the cellular and viral membranes, followed by virus-cell membrane fusion.
In coronavirus spike proteins, S1 is mainly responsible for binding to receptor and S2 for fusion (5). The flexibility between HR1 and HR2 is not sufficient enough to allow for complete conformational changes to occur, although the two heptad repeat regions have a great tendency to pack closely together. Instead, other intervening regions of the spike protein should promote the formation of the fusion core. Further experiments should reveal how the exact conformational change occurs and how best to inhibit the viral fusion.
Potential Inhibitors of Coronaviruses Entry into Cells—The sequence alignment of HR1 and HR2 regions in spike proteins among coronaviruses shows significant similarity in their heptad repeat regions (Fig. 1B). For example, between MHV and SARS, identity of HR1 is 60% and positive is 91%, identity of HR2 is 35 and positive is 85%. Important residues for six-helix bundle formation, located in the a, d, e, and g positions of HR1 and a and d positions of HR2, are all highly conserved. We can conclude that coronavirus spike proteins share a similar binding region of the HR1·HR2 complex and three-dimensional fusion core structure. Analogous to the HIV C-peptides, the HR2 region of coronavirus spike proteins most likely functions in a dominant-negative manner by binding to the transiently exposed hydrophobic grooves in the prehairpin intermediate and consequently blocking the formation of the fusion-active hairpin structures (43). These strategies have been used successfully in fusion inhibitors design for HIV (7, 42, 49, 50, 51). A similar approach may be applied to identify inhibitors of coronavirus infection. HR1 and HR2 regions and their derivatives are all potential inhibitors. Cavities and grooves on the surface of the central coiled coil are strong potential binding sites for small molecule inhibitors.
In conclusion, the crystal structure of MHV fusion core shows the first fusion core structure of any coronavirus. Although the structure shares common features with those of other viral fusion proteins, it has unique characteristics that distinguish it from other fusion core structures. Sequence alignment of HR regions among coronaviruses indicates a similar structure among coronavirus spike proteins and suggests a common mechanism for viral fusion. This structure will also open an avenue toward the structure-based fusion inhibitor design of peptides, or peptide analogs, e.g. small molecules, targeted against emerging infectious diseases, such as SARS.
Acknowledgments
We thank Dr. Mark Bartlam for comments and critical reading.
Footnotes
The atomic coordinates and structure factors (codes 1WDF and 1WDG) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
The abbreviations used are: MHV, mouse hepatitis virus; HA, hemagglutinin; HIV, human immunodeficiency virus; HR, heptad repeat; MES, 4-morpholineethanesulfonic acid; MMLV, Moloney murine leukemia virus; S protein, spike protein; SARS, severe acute respiratory syndrome; SIV, simian immunodeficiency virus; TM, transmembrane.
Y. Xu, Z. Bai, L. Qin, X. Li, G. F. Gao, and Z. Rao, submitted for publication.
References
- 1.Spaan W., Cavanagh D., Horzinek M.C. J. Gen. Virol. 1988;69:2939–2952. doi: 10.1099/0022-1317-69-12-2939. [DOI] [PubMed] [Google Scholar]
- 2.Lee H.J., Shieh C.K., Gorbalenya A.E., Koonin E.V., La Monica N., Tuler J., Bagdzhadzhyan A., Lai M.M. Virology. 1991;180:567–582. doi: 10.1016/0042-6822(91)90071-I. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siddell S., Wege H., Ter Meulen V. J. Gen. Virol. 1983;64:761–776. doi: 10.1099/0022-1317-64-4-761. [DOI] [PubMed] [Google Scholar]
- 4.Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P., Penaranda S., Bankamp B., Maher K., Chen M.H., Tong S., Tamin A., Lowe L., Frace M., DeRisi J.L., Chen Q., Wang D., Erdman D.D., Peret T.C., Burns C., Ksiazek T.G., Rollin P.E., Sanchez A., Liffick S., Holloway B., Limor J., McCaustland K., Olsen-Rasmussen M., Fouchier R., Gunther S., Osterhaus A.D., Drosten C., Pallansch M.A., Anderson L.J., Bellini W.J. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 5.Gallagher T.M., Buchmeier M.J. Virology. 2001;279:371–374. doi: 10.1006/viro.2000.0757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davies H.A., Macnaughton M.R. Arch. Virol. 1979;59:25–33. doi: 10.1007/BF01317891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eckert D.M., Kim P.S. Annu. Rev. Biochem. 2001;70:777–810. doi: 10.1146/annurev.biochem.70.1.777. [DOI] [PubMed] [Google Scholar]
- 8.Hernandez L.D., Hoffman L.R., Wolfsberg T.G., White J.M. Annu. Rev. Cell Dev. Biol. 1996;12:627–661. doi: 10.1146/annurev.cellbio.12.1.627. [DOI] [PubMed] [Google Scholar]
- 9.Yamada Y.K., Takimoto K., Yabe M., Taguchi F. Virology. 1997;227:215–219. doi: 10.1006/viro.1996.8313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wool-Lewis R.J., Bates P. J. Virol. 1999;73:1419–1426. doi: 10.1128/jvi.73.2.1419-1426.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hernandez L.D., Peters R.J., Delos S.E., Young J.A., Agard D.A., White J.M. J. Cell Biol. 1997;139:1455–1464. doi: 10.1083/jcb.139.6.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Damico R.L., Crane J., Bates P. Proc. Natl. Acad. Sci. U. S. A. 1998;95:2580–2585. doi: 10.1073/pnas.95.5.2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chambers P., Pringle C.R., Easton A.J. J. Gen. Virol. 1990;71:3075–3080. doi: 10.1099/0022-1317-71-12-3075. [DOI] [PubMed] [Google Scholar]
- 14.Bullough P.A., Hughson F.M., Skehel J.J., Wiley D.C. Nature. 1994;371:37–43. doi: 10.1038/371037a0. [DOI] [PubMed] [Google Scholar]
- 15.Lu M., Blacklow S.C., Kim P.S. Nat. Struct. Biol. 1995;2:1075–1082. doi: 10.1038/nsb1295-1075. [DOI] [PubMed] [Google Scholar]
- 16.Weissenhorn W., Dessen A., Harrison S.C., Skehel J.J., Wiley D.C. Nature. 1997;387:426–430. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- 17.Blacklow S.C., Lu M., Kim P.S. Biochemistry. 1995;34:14955–14962. doi: 10.1021/bi00046a001. [DOI] [PubMed] [Google Scholar]
- 18.Weissenhorn W., Wharton S.A., Calder L.J., Earl P.L., Moss B., Aliprandis E., Skehel J.J., Wiley D.C. EMBO J. 1996;15:1507–1514. [PMC free article] [PubMed] [Google Scholar]
- 19.Chan D.C., Fass D., Berger J.M., Kim P.S. Cell. 1997;89:263–273. doi: 10.1016/s0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- 20.Tan K., Liu J., Wang J., Shen S., Lu M. Proc. Natl. Acad. Sci. U. S. A. 1997;94:12303–12308. doi: 10.1073/pnas.94.23.12303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caffrey M., Cai M., Kaufman J., Stahl S.J., Wingfield P.T., Covell D.G., Gronenborn A.M., Clore G.M. EMBO J. 1998;17:4572–4584. doi: 10.1093/emboj/17.16.4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weissenhorn W., Carfi A., Lee K.H., Skehel J.J., Wiley D.C. Mol. Cell. 1998;2:605–616. doi: 10.1016/s1097-2765(00)80159-8. [DOI] [PubMed] [Google Scholar]
- 23.Weissenhorn W., Calder L.J., Wharton S.A., Skehel J.J., Wiley D.C. Proc. Natl. Acad. Sci. U. S. A. 1998;95:6032–6036. doi: 10.1073/pnas.95.11.6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baker K.A., Dutch R.E., Lamb R.A., Jardetzky T.S. Mol. Cell. 1999;3:309–319. doi: 10.1016/s1097-2765(00)80458-x. [DOI] [PubMed] [Google Scholar]
- 25.Wiley D.C., Skehel J.J. Annu. Rev. Biochem. 1987;56:365–394. doi: 10.1146/annurev.bi.56.070187.002053. [DOI] [PubMed] [Google Scholar]
- 26.Wilson I.A., Skehel J.J., Wiley D.C. Nature. 1981;289:366–373. doi: 10.1038/289366a0. [DOI] [PubMed] [Google Scholar]
- 27.Carr C.M., Kim P.S. Cell. 1993;73:823–832. doi: 10.1016/0092-8674(93)90260-w. [DOI] [PubMed] [Google Scholar]
- 28.Bosch B.J., van der Zee R., de Haan C.A., Rottier P.J. J. Virol. 2003;77:8801–8811. doi: 10.1128/JVI.77.16.8801-8811.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Singh M., Berger B., Kim P.S. J. Mol. Biol. 1999;290:1031–1041. doi: 10.1006/jmbi.1999.2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Otwinowski Z., Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 31.Terwilliger T.C., Berendzen J. Acta Crystallogr. Sect. D Biol. Crystallogr. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Terwilliger T.C. Acta Crystallogr. Sect. D Biol. Crystallogr. 2001;57:1755–1762. doi: 10.1107/S0907444901013737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones T.A., Zou J.Y., Cowan S.W., Kjeldgaard Acta Crystallogr. Sect. A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 34.Laskowski R.A., MacArthur M.W., Moss D.S., Thornton J.M. J. Appl. Crystallogr. 1993;26:283–291. [Google Scholar]
- 35.Nicholls A., Sharp K.A., Honig B. Proteins. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- 36.Guex N., Peitsch M.C. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 37.Kraulis P.J. J. Appl. Crystallogr. 1991;24:946–950. [Google Scholar]
- 38.Brunger A.T., Adams P.D., Clore G.M., DeLano W.L., Gros P., Grosse-Kunstleve R.W., Jiang J.S., Kuszewski J., Nilges M., Pannu N.S., Read R.J., Rice L.M., Simonson T., Warren G.L. Acta Crystallogr. Sect. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 39.Zhao X., Singh M., Malashkevich V.N., Kim P.S. Proc. Natl. Acad. Sci. U. S. A. 2000;97:14172–14177. doi: 10.1073/pnas.260499197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fass D., Kim P.S. Curr. Biol. 1995;5:1377–1383. doi: 10.1016/s0960-9822(95)00275-2. [DOI] [PubMed] [Google Scholar]
- 41.Malashkevich V.N., Schneider B.J., McNally M.L., Milhollen M.A., Pang J.X., Kim P.S. Proc. Natl. Acad. Sci. U. S. A. 1999;96:2662–2667. doi: 10.1073/pnas.96.6.2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wild C.T., Shugars D.C., Greenwell T.K., McDanal C.B., Matthews T.J. Proc. Natl. Acad. Sci. U. S. A. 1994;91:9770–9774. doi: 10.1073/pnas.91.21.9770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chan D.C., Kim P.S. Cell. 1998;93:681–684. doi: 10.1016/s0092-8674(00)81430-0. [DOI] [PubMed] [Google Scholar]
- 44.Luo Z., Weiss S.R. Virology. 1998;244:483–494. doi: 10.1006/viro.1998.9121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Luo Z., Matthews A.M., Weiss S.R. J. Virol. 1999;73:8152–8159. doi: 10.1128/jvi.73.10.8152-8159.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Modis Y., Ogata S., Clements D., Harrison S.C. Nature. 2004;427:313–319. doi: 10.1038/nature02165. [DOI] [PubMed] [Google Scholar]
- 47.Gibbons D.L., Vaney M.C., Roussel A., Vigouroux A., Reilly B., Lepault J., Kielian M., Rey F.A. Nature. 2004;427:320–325. doi: 10.1038/nature02239. [DOI] [PubMed] [Google Scholar]
- 48.Han X., Bushweller J.H., Cafiso D.S., Tamm L.K. Nat. Struct. Biol. 2001;8:715–720. doi: 10.1038/90434. [DOI] [PubMed] [Google Scholar]
- 49.Eckert D.M., Kim P.S. Proc. Natl. Acad. Sci. U. S. A. 2001;98:11187–11192. doi: 10.1073/pnas.201392898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eckert D.M., Malashkevich V.N., Hong L.H., Carr P.A., Kim P.S. Cell. 1999;99:103–115. doi: 10.1016/s0092-8674(00)80066-5. [DOI] [PubMed] [Google Scholar]
- 51.Root M.J., Kay M.S., Kim P.S. Science. 2001;291:884–888. doi: 10.1126/science.1057453. [DOI] [PubMed] [Google Scholar]