

Abstract
We investigated a role for endogenous Rap1, a small monomeric GTP-binding protein of the Ras family, in nerve growth factor (NGF) signaling in PC12 cells. Although both epidermal growth factor (EGF) and NGF caused transient activation of Ras, only NGF induced the activation of Rap1. Moreover, Rap1 activation was sustained for hours, an effect that matched the sustained activation of the mitogen-activated protein kinase (MAPK) pathway. To investigate the molecular basis for Rap1 activation, we examined complexes containing C3G, a guanine nucleotide exchange factor for Rap1, and CrkL, an adapter protein known to influence Rap1 signaling. NGF induced the formation of a long-lived complex containing C3G/CrkL/Shp2/Gab2/TrkA. Linking the complex to Rap1 activation, we coprecipitated activated TrkA and activated MAPK with activated Rap1 in NGF-treated cells. Confocal microscopy and subcellular fractionation showed that activated Rap1 and the other proteins of the signaling complex were present in endosomes. Pretreatment of PC12 cells with brefeldin A (BFA), which disrupts the Golgi and endosomal compartments, had little effect on Ras activation but strongly inhibited NGF-induced Rap1 activation and continuing MAPK activation. We propose that endosomes are a site from which NGF induces the prolonged activation of Rap1 and MAPK.
Keywords: PC12, NGF, TrkA, Gab2, Rap1, endosome
Nerve growth factor (NGF) is a target-released factor that promotes the survival and differentiation of responsive neurons (Meakin and Shooter, 1992; Chao et al., 1998;Howe and Mobley, 2001). The signal that is initiated by NGF at the axonal terminal must move retrogradely to elicit NGF actions in the cell body. Because in some neurons the NGF signal must move over long distances, the signal must be specific, robust, and long-lived. Recent studies suggest that endosomes containing NGF and activated TrkA may signal with these properties (Grimes et al., 1996, 1997). Also, it is known that complexes containing endocytosed NGF and activated TrkA are transported retrogradely from the axon to the cell body (Ehlers et al., 1995; Riccio et al., 1997; Sandow et al., 2000) and that the retrogradely transported signal results in long-lasting activation of cAMP response element binding proteins (CREB; Riccio et al., 1997,1999). The findings suggest that retrograde NGF signaling could be mediated by endosomes.
NGF signals via the mitogen-activated protein kinase (MAPK) pathway to influence many aspects of neuronal survival and differentiation. Persistent activation of the MAPK pathway has been postulated to be essential for NGF-induced differentiation of pheochromocytoma (PC12) cells (Boulton et al., 1991; Cowley et al., 1994; Young et al., 1994;Marshall, 1995; Pang et al., 1995). Because many of the differentiative responses to MAPK are mediated in the neuronal cell body, we hypothesized that signaling endosomes are a source of persistent MAPK signaling. To explore the mechanism by which NGF induces persistent MAPK activation, we tested the involvement of Rap1. Recent attention has focused on the signaling properties of this protein, a member of the Ras family (Bos, 1998; Grewal et al., 1999). Studies using transient transfection demonstrated that NGF activated His-tagged Rap1 to stimulate the MAPK pathway via the B-Raf kinase (York et al., 1998). On the basis of these results, Stork and colleagues proposed that Rap1 activates B-Raf to cause sustained activation of the Mek/MAPK cascade (York et al., 1998; Grewal et al., 1999). However, Zwartkruis et al. (1998) disputed this interpretation because they failed to detect the activation of endogenous Rap1 in NGF-treated PC12 cells. To reexamine the issue and to circumvent possible artifacts attributable to microinjection and transfection/overexpression, we analyzed NGF effects on endogenous Ras and Rap1 signaling in PC12 cells.
We demonstrated that, whereas both NGF and epidermal growth factor (EGF) induced transient Ras activation, only NGF elicited the stimulation of Rap1. Persistent Rap1 activation correlated temporally with the continuing activation of MAPK. C3G and CrkL, proteins shown to mediate signaling to Rap1, were present in signaling protein complexes in cells that were treated with NGF and EGF, but the composition of these complexes differed markedly. In NGF-treated cells a long-lived complex that contained C3G, CrkL, Shp2, Gab2, and TrkA was present. Compelling evidence for a significant role for Rap1 in NGF signaling was the finding that activated Rap1 specifically could be precipitated together with activated TrkA and activated MAPK in NGF-treated cells. To establish the cellular site of Rap1 signaling, we performed subcellular fractionation and confocal microscopic studies. Both showed that after NGF treatment Rap1 was present together with the other proteins of the signaling complex and with activated MAPK in endosomes. By treating cells with brefeldin A (BFA) to disrupt the Golgi/endosomal compartments, we specifically inhibited Rap1 activation and blocked persistent MAPK activation. Our results are evidence that NGF signals via Rap1 in endosomes to activate the MAPK pathway persistently.
MATERIALS AND METHODS
Antibodies and reagents. Mouse antibodies to EEA1, Ras, and Rap1 were purchased from Transduction Laboratories (Lexington, KY). Rabbit antibodies to the EGF receptor (EGFR), Mek1, Rab5B, Rap1 (Krev-1), C3G, Cbl, CrkL, B-Raf, Shp2, and a mouse antibody to TrkA (MCTrks) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse antibodies to phosphotyrosine (4G10) and Erk2 (MAPK2) were purchased from Upstate Biotechnology (Lake Placid, NY). Rabbit antibodies to pTrkA (pY490), MAPK1/2 (Erk1/2), and the phosphorylated, activated MAPK1/2 were purchased from New England Biolabs (Beverly, MA). Rabbit antisera against Gab2 were a kind gift from Dr. G.-S. Feng (Indiana University School of Medicine, IN). BFA, K252a,n-octyl-β-d-glucopyranoside, EGF, and basic fibroblast growth factor (bFGF) were purchased from CalBiochem-NovaBiochem (San Diego, CA). Mouse NGF was purified as described previously (Mobley et al., 1986). Horseradish peroxidase (HRP) conjugated to goat anti-rabbit or anti-mouse IgGs was obtained from Jackson ImmunoResearch Laboratories (West Grove, PA). Alexa 568 goat anti-mouse IgG and Alexa 488 goat anti-rabbit IgG conjugates were purchased from Molecular Probes (Eugene, OR). Protein A–agarose conjugates and SuperSignal reagents were obtained from Pierce (Rockford, IL). OptiPrep was obtained from Life Technologies (Rockville, MD). Agarose–glutathione conjugates and all other chemicals were purchased from Sigma (St. Louis, MO).
Cell culture. PC12 and 6-24 cells (a PC12 cell line that overexpresses the wild-type TrkA receptor) (Stephen et al., 1994) were maintained in DMEM (4.5 mg/l glucose) supplemented with 10% heat-inactivated horse serum and 5% fetal bovine serum (FBS). Cells were incubated at 37°C with 5% CO2. Cells that were 50–60% confluent and serum-starved for 24 hr before treatments were used in all experiments. All treatments were performed by adding growth factors and other reagents to the media at 37°C.
Ras and Rap1 activation assay. Established methods were used to detect endogenous GTP-bound Ras and Rap1 proteins (Herrmann et al., 1995, 1996). The fusion constructs between glutathioneS-transferase (GST) and either the Rap binding domain of RalGDS (RalGDSRBD) or the Ras binding domain of C-Raf (C-RafRBD) (gifts from Dr. J. L. Bos, Utrecht University, Netherlands) were overexpressed in Escherichia coli DH5α cells. The fusion proteins were purified (Herrmann et al., 1995) and used to assay for Rap1GTP and RasGTP, respectively. Briefly, equal numbers of treated or untreated PC12 cells were lysed on ice for 30 min in fishing buffer [FB; 10% glycerol, 1% NP-40, and (in mm) 50 Tris-HCl, pH 7.5, 200 NaCl, 2.0 MgCl2, 10 NaF, and 1 Na3VO4 plus 250 μm phenylmethylsulfonyl fluoride (PMSF), 2 μg/ml aprotinin, 1 μg/ml leupeptin, and 10 μg/ml soybean trypsin inhibitor]. The samples were centrifuged at 14,000 rpm for 30 min at 4°C. Either 5 μg of RalGDSRBD/GST or 5 μg of C-RafRBD/GST proteins prebound to agarose–glutathione conjugates was added to the resulting supernatants and incubated at 4°C for 60–120 min with gentle rotation. The beads were washed four times in cold FB and boiled in SDS-PAGE sample buffer. The amounts of RasGTP and Rap1GTP were analyzed by SDS-PAGE and immunoblotting.
Cell fractionation. Fractionation of PC12 cells was performed at 4°C, using a slight modification of the method described by Sheff et al. (1999). Equal numbers of treated or untreated PC12 cells were rinsed and harvested by centrifugation (800 ×g for 5 min). Cells were resuspended and homogenized by douncing 20 times in a Teflon-coated homogenizer in 1 ml of cold homogenization buffer [HB; (in mm) 250 sucrose, 20 Tricine-NaOH, pH7.8, 1 EDTA, and 2 MgCl2]. The samples were centrifuged (800 × g for 10 min), and the supernatant was adjusted to 25% OptiPrep with 50% OptiPrep in HB. The resulting mixture (2 ml in 25% OptiPrep) was placed at the bottom of an Ultra-Clear Tube (14 × 89 mm; Beckman Instruments, Palo Alto, CA) and was overlaid successively with 2 ml each of 20, 15, 10, and 5% OptiPrep in cold HB. The samples were centrifuged for 16–18 hr at 27,000 rpm at 4°C in a SW41 rotor (Beckman Instruments). Membrane fractions were collected from each of the four interphases of the OptiPrep gradients. For Rap1GTP assay the membrane fractions were adjusted to 1× FB by adding 3× FB to the sample. Then the Rap1GTP assay was performed as described above. For some studies, proteins were precipitated from these membrane fractions by using 7% trichloroacetic acid (TCA) and were washed with acetone. The pellets were air-dried, boiled in SDS-PAGE loading buffer, and analyzed by SDS-PAGE and immunoblotting.
Immunoprecipitation, SDS-PAGE, and immunoblotting. Equal numbers of treated or untreated cells were washed and lysed in the TET buffer (25 mm Tris-HCl, pH7.5, 5 mm EDTA, 1% Triton X-100) containing 150 mm NaCl, 60 mmn-octyl-β-d-glucopyranoside, 250 μm PMSF, and 1 mmNa3VO4. The samples were centrifuged, and the resulting supernatants were incubated overnight at 4°C with 4 μg of rabbit IgGs against C3G or CrkL with gentle rotation. The immunocomplexes were collected by using protein A–agarose conjugates. All immunoprecipitates were washed twice in cold TET containing 250 mm NaCl and twice with cold 10 mm Tris-HCl, pH 7.5, containing 5 mm EDTA and were boiled in SDS-PAGE sample buffer. The samples were separated on 7.5–12.5% gels, and proteins were electrotransferred onto polyvinylidene difluoride (PVDF) membranes. The PVDF membranes were preblotted with 5% nonfat milk (Carnation) and probed with primary antibodies, as indicated, at concentrations suggested by the suppliers. The blots were washed in TBST (0.1% Tween 20 in TBS), followed by incubation with either goat anti-mouse or anti-rabbit IgG–HRP conjugates at a dilution of 1:10,000–1:40,000. The blots were washed and developed with SuperSignal (Pierce).
Indirect immunofluorescence. PC12 cells were grown for 24–48 hr on glass coverslips coated with Matrigel (Becton Dickinson, San Diego, CA). Cells were washed twice briefly with cold PBS and fixed with ice-cold 100% methanol for 5 min. The samples were rinsed three times with cold 50% methanol, followed by three rinses with PBS at room temperature (RT). The fixed cells were preblocked with 0.8% bovine serum albumin (BSA) in PBS for 20 min at RT. Primary antibodies were diluted at 1:300–1:500 in PBS containing 0.8% BSA and were incubated with the fixed cells for 1 hr at RT. The samples were washed with PBS containing 0.8% BSA three times and then incubated with goat anti-rabbit or anti-mouse–IgG Alexa conjugates (1:600) for 1 hr at RT. The coverslips were washed with PBS containing 0.2% BSA three times, followed by a rinse with PBS and dH2O. The coverslips were air-dried and mounted in anti-fade medium for observation by confocal microscopy. The images were captured with a Nikon Eclipse E800 microscope and a Bio-Rad Laser-Scanning System Radiance2000 (Hercules, CA). Using a 60× Plan-Apo immersion objective (numerical aperture, 1.4), we collected images at the half-height of the cell with an iris of 2.0 mm, a zoom of 3, five passes with a Kalman filter, and a 512 × 512 collection box. The images were processed by Confocal Assistant 4.02 (Bio-Rad) and Adobe Photoshop 5.0 (Adobe Systems, Mountain View, CA).
RESULTS
NGF, but not EGF, stimulated prolonged activation of MAPK
To examine the signaling pathways activated by EGF and NGF, we first evaluated their effects on MAPK activation in PC12 cells. Serum-starved cells were treated with either EGF (50 ng/ml) or NGF (50 ng/ml) for the indicated time intervals (Fig.1A). Cell lysates were analyzed by SDS-PAGE and immunoblotting. EGF induced the transient activation of MAPK, as detected with an antibody that specifically recognizes the phosphorylated (i.e., activated) forms of MAPK (Fig.1A). Activated MAPK was seen at 2–5 min but was not detected after 5 min. NGF-stimulated MAPK activation showed a different time course. Activated MAPK was detected at 2 min and reached the maximal level at 5 min. However, in contrast to EGF treatment, significant MAPK activation was still detected at 60 min of NGF treatment (Fig. 1A). As a loading control, we reprobed the blot with an antibody to MAPK2, the 42 kDa MAPK. The total amount of MAPK2 was unchanged during the experiment. We conclude that NGF stimulates the sustained activation of MAPK.
Fig. 1.

Analysis of endogenous Ras and Rap1 activation in EGF- and NGF-treated cells. Equal numbers of serum-starved PC12 cells were treated with either EGF (50 ng/ml) or NGF (50 ng/ml) for the indicated time intervals or the vehicle control (0 min).A, The cells were rinsed and lysed in RIPA buffer. Proteins (40 μg) from each cell lysate were separated on SDS-PAGE and analyzed by immunoblotting. The blot was probed with a rabbit antibody that specifically recognizes phosphorylated MAPK (top). Then the blot was reprobed with a mouse antibody against MAPK2 (bottom). B, RasGTPwas precipitated with the C-RafRBD/GST fusion protein. The fusion protein was prebound to glutathione–agarose beads before incubation with cell lysates. After incubation the beads were washed and boiled in SDS-PAGE sample buffer. The samples were separated on 12.5% SDS-PAGE, and the proteins were transferred onto PVDF membrane. The membrane was probed with a mouse antibody to Ras. C, The amounts of Rap1GTP that followed EGF and NGF treatment were assayed as in B, except that the RalGDSRBD/GST fusion proteins were used to precipitate Rap1GTP. The resulting blot was probed with a mouse antibody to Rap1.D, Cells were pretreated with either vehicle (−) or 0.5 μm K252a (+) for 15 min at 37°C before treatment with NGF (50 ng/ml) for the indicated times. Activated Rap1 was assayed as in C. E, Serum-starved 6-24 cells were treated with NGF (50 ng/ml) for the indicated time intervals or the vehicle control (0 min). The amount of Rap1GTP was assayed as in C. All blots were visualized with SuperSignal; the results shown are representative of at least three independent experiments.
NGF and EGF induced transient activation of endogenous Ras in PC12 cells
To ask whether the activation of endogenous Ras could account for the sustained activation of MAPK elicited by NGF, we examined Ras activation via an established method (Herrmann et al., 1995). The method takes advantage of specific binding of a C-RafRBD/GST fusion protein to the activated form of Ras (i.e., RasGTP). The amount of RasGTP that followed EGF or NGF treatment was analyzed by SDS-PAGE and immunoblotting. Representative blots are shown in Figure 1B. Ras was activated rapidly by both EGF and NGF (Fig. 1B). EGF-induced Ras activation was detected as early as ∼10 sec, reached the maximal level at ∼2 min, decreased to a low level at 5 min, and was detected only weakly thereafter (Fig. 1B). A similar pattern for Ras activation was observed with NGF treatment. NGF-induced Ras activation was maximal at 2 min but was no longer detected after 5 min (Fig.1B). Thus, the transient activation of endogenous Ras alone is insufficient for the prolonged activation of MAPK in NGF-treated cells.
Rap1 was activated by NGF, but not by EGF
We next investigated whether endogenous Rap1 was activated by NGF. Similar to the assay used for activated Ras, a RalGDS-RBD/GST fusion protein was used to analyze Rap1 activation (Herrmann et al., 1996). No significant activation of Rap1 was detected after EGF treatment (Fig.1C). In contrast, NGF treatment elicited a marked increase in Rap1GTP (Fig. 1C). Moreover, unlike the transient pattern seen for Ras activation (Fig.1B), NGF-induced Rap1 activation was persistent. Although Rap1 was activated weakly at early time points (2–10 min; Fig. 1C), there was a marked increase at 30 min, and the activation continued through at least 180 min. The increase at 30 min and thereafter was not attributable to newly synthesized Rap1, because we did not observe an increase in Rap1 protein by immunoblotting, and cycloheximide, an inhibitor of protein synthesis, had no effect (data not shown).
To investigate further the NGF effect on Rap1, we tested 6-24 cells, a PC12 cell line that overexpresses TrkA (Stephen et al., 1994). The 6-24 cells were treated with NGF (50 ng/ml) for the indicated time intervals. Activated Rap1 was assayed as described above. Our results showed that NGF treatment activated Rap1 at 5 min. There was a marked increase in Rap1 activation after 10 min. The maximal level was seen at 60–180 min (Fig. 1E). Because Rap1 was activated more robustly in cells that overexpress TrkA, the possibility was raised that NGF signals via TrkA to induce Rap1 activation.
To show that TrkA was required for NGF-induced persistent Rap1 activation, we pretreated serum-starved PC12 cells with either vehicle or 0.5 μm of K252a, a TrkA-specific inhibitor, before NGF treatment. Activation of Rap1 was assayed as described previously. The results showed that pretreatment with K252a abolished NGF-induced Rap1 activation (Fig. 1D). Thus, we conclude that NGF acts via TrkA to induce the persistent activation of Rap1 and that this effect correlates temporally with the sustained activation of MAPK in NGF-treated cells.
bFGF also stimulated Rap1 activation and MAPK in PC12 cells
Like NGF, bFGF induces the differentiation of PC12 cells (Greene and Tischler, 1982). We reasoned that if Rap1 activation and the persistent activation of MAPK are both required for the differentiation of PC12 cells, bFGF treatment should result in the induction of both responses. We tested the effect of bFGF on the activation of Ras, Rap1, and MAPK. When serum-starved PC12 cells were treated with bFGF (50 ng/ml), Ras activation was induced transiently in a pattern similar to that for NGF (Fig.2A). Ras was activated maximally at 2–5 min and was downregulated rapidly thereafter (Fig.2A). Activation of Rap1 was detected as early as 5 min, and the maximal level was seen at 60 min (Fig.2B). Similar to treatment with NGF, activated Rap1 was still detected at 180 min in bFGF-treated cells (Fig.2B). As seen with NGF, bFGF induced the sustained activation of MAPK (Fig. 2C). Activated MAPK was detected at 2 min and persisted through at least 60 min. In demonstrating that the prolonged activation of both Rap1 and MAPK was induced by bFGF, our findings lend further support to the suggested links among the activation of endogenous Rap1, the persistent activation of MAPK, and the differentiation of PC12 cells.
Fig. 2.

bFGF caused persistent activation of endogenous Rap1 and MAPK in PC12 cells. Equal numbers of serum-starved PC12 cells were treated with bFGF (50 ng/ml) for the indicated time intervals or the vehicle control (0 min), and activation of Ras (A) or Rap1 (B) was assayed as in Figure 1. C, Equal numbers of serum-starved PC12 cells were treated with bFGF (50 ng/ml) for the indicated time intervals or the vehicle control (0 min). Cells were rinsed and then lysed in RIPA buffer as in Figure 1. Proteins (40 μg) from each cell lysate were separated on SDS-PAGE and analyzed by immunoblotting. The blot was probed with a rabbit antibody against phosphorylated MAPK. Then the blot was reprobed with a rabbit antibody to total MAPK. All blots were visualized with SuperSignal; the results shown are representative of at least three independent experiments.
NGF and EGF differentially affected C3G, the Rap1-specific guanine nucleotide exchange factor
To investigate how NGF induced the persistent activation of Rap1, we examined the effect of NGF treatment on signaling proteins that regulate the activity of Rap1. The results for NGF were compared with those for EGF, because EGF failed to activate Rap1. C3G is a guanine nucleotide exchange factor that specifically interacts with and activates Rap1 by catalyzing the conversion of Rap1 from the inactive (Rap1GDP) to the active form (Rap1GTP; Gotoh et al., 1995). Because tyrosine phosphorylation has been suggested to be required for C3G activity (Ichiba et al., 1999), we evaluated the tyrosine phosphorylation state of C3G. Serum-starved PC12 cells were treated with either EGF or NGF for time intervals ranging from 0 to 30 min. C3G was immunoprecipitated from cell lysates and analyzed by SDS-PAGE and immunoblotting. C3G was tyrosine phosphorylated constitutively in PC12 cells, as revealed by blotting with 4G10 (Fig.3A,B). EGF treatment induced the rapid dephosphorylation of C3G and caused the rapid association of C3G with a number of tyrosine-phosphorylated proteins for which the estimated molecular masses ranged from 95 to 170 kDa (Fig.3A). This observation suggests that tyrosine phosphorylation may regulate the ability of C3G to interact with other proteins. In contrast to the findings for EGF, NGF treatment did not produce an appreciable change in tyrosine-phosphorylated C3G, nor was C3G shown to become associated with novel tyrosine-phosphorylated proteins in NGF-treated cells (Fig. 3B). To investigate further the presence of tyrosine phosphorylation of C3G after EGF or NGF treatment, we immunoprecipitated tyrosine-phosphorylated proteins with anti-phosphotyrosine antibodies (4G10) from cells that were treated with either EGF or NGF for time intervals ranging from 0 to 30 min. The precipitates were separated on SDS-PAGE and immunoblotted with anti-C3G antibodies. Our results confirmed that C3G became dephosphorylated transiently and markedly in EGF-treated cells (Fig. 3C). No other bands corresponding to tyrosine-phosphorylated C3G were evident in EGF-treated cells at 2 min (Fig. 3C). In NGF-treated cells the level of tyrosine phosphorylation in C3G was somewhat decreased relative to the control, but tyrosine-phosphorylated C3G was readily evident at all time points.
Fig. 3.
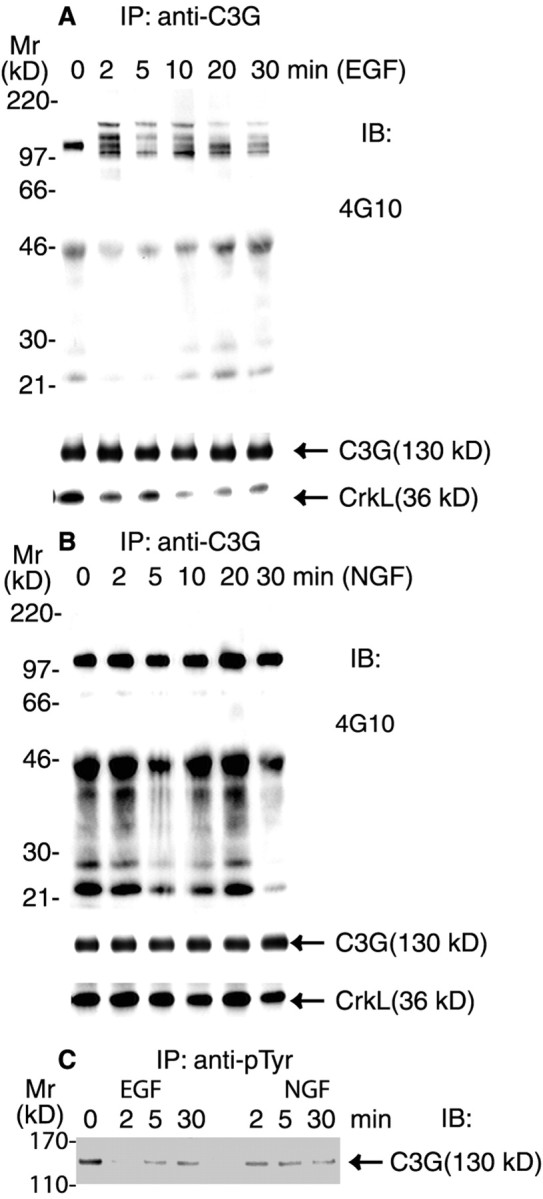
C3G was tyrosine phosphorylated constitutively and formed a complex with CrkL in PC12 cells. Equal numbers of serum-starved PC12 cells were treated with EGF (50 ng/ml;A) and NGF (50 ng/ml; B) for the indicated time intervals or the vehicle control (0 min). Cells were washed and then lysed as described in Materials and Methods. The supernatants were incubated with 4 μg of rabbit IgGs against C3G (A, B). C, Equal numbers of serum-starved PC12 cells were treated with EGF (50 ng/ml) or NGF (50 ng/ml) for the indicated time intervals or the vehicle control (0 min). Cells were washed and then lysed. The supernatants were incubated with 4 μg of mouse IgGs against phosphotyrosine (4G10). All immunoprecipitates were analyzed by 10% SDS-PAGE and immunoblotting. The resulting blots were probed with the specified antibody. All blots were visualized with SuperSignal; the results shown are representative of at least three independent experiments.
In several cell lines C3G has been shown to form complexes with Crk in response to growth factor stimulation (Gotoh et al., 1995; Boussiotis et al., 1997; Okada and Pessin, 1997). We asked which of the two predominant Crk isoforms, CrkII or CrkL, was present in the C3G immunoprecipitates. The 40 kDa CrkII was not detected in the complex by immunoblotting (data not shown). The 36 kDa CrkL was found to be associated constitutively with C3G in serum-starved PC12 cells (Fig.3A,B). EGF treatment resulted in a marked decrease in the amount of CrkL (Fig. 3A). The decrease was apparent by 2 min and persisted through 30 min. NGF treatment did not result in a decrease in CrkL in complex with C3G (Fig. 3B). These results demonstrate clear differences for EGF and NGF signaling on tyrosine phosphorylation of C3G and on proteins found in complex with C3G.
NGF and EGF induced the formation of different multicomponent signaling protein complexes containing C3G and CrkL
CrkL has been shown to play an important role in regulating Rap1 activation and in suppressing Ras-dependent signaling in T lymphocytes (Boussiotis et al., 1997). When cotransfected with C3G, CrkL was effective in enhancing Rap1 activation in PC12 cells (York et al., 1998). These findings together with our data for the continued association of C3G and CrkL after NGF treatment pointed to the possibility that endogenous CrkL might regulate NGF signaling to Rap1. To examine a role for CrkL, we treated serum-starved PC12 cells with either EGF or NGF. CrkL was immunoprecipitated and analyzed by SDS-PAGE and immunoblotting (Fig. 4). A short exposure of the blots probed with 4G10 showed that several tyrosine-phosphorylated proteins were associated with CrkL in unstimulated cells. There were at least three such prominent proteins with apparent molecular masses of 130, 95, and 68 kDa (Fig.4A). None of these proteins was detected after 5 min of EGF treatment. Instead, a new protein with a molecular mass of 120 kDa appeared in the CrkL immunoprecipitates in EGF-treated cells (Fig.4A). At 10 min the tyrosine-phosphorylated proteins seen in the quiescent state were detected again. A longer exposure of Figure 4A revealed additional differences between EGF and NGF treatments (Fig. 4B). EGF induced the transient association of CrkL with a 170 kDa tyrosine-phosphorylated protein (Fig. 4B). In contrast, NGF treatment induced the appearance of a tyrosine-phosphorylated 84 kDa protein that persisted in the CrkL complexes through 60 min (Fig.4B, arrow). We conclude that CrkL forms complexes with different proteins in response to EGF and NGF treatments.
Fig. 4.
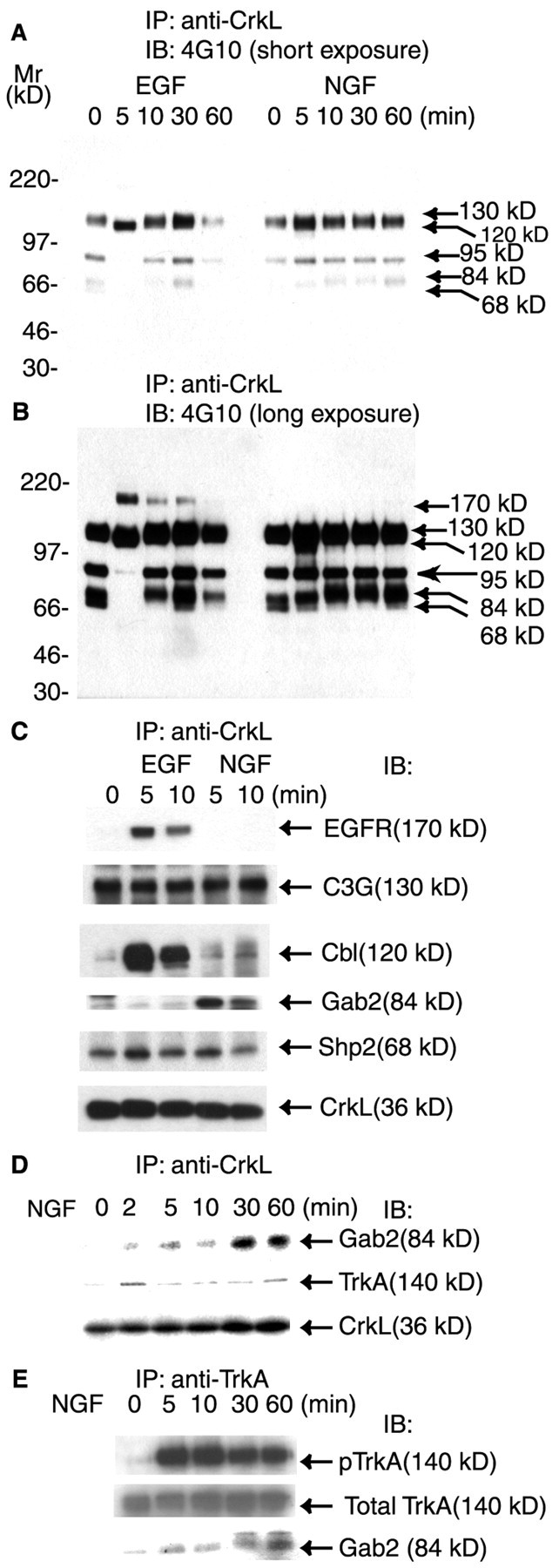
Different CrkL-containing complexes were induced by NGF and EGF treatment. Equal numbers of serum-starved PC12 cells were treated with either EGF (50 ng/ml) or NGF (50 ng/ml) for the indicated time intervals or the vehicle control (0 min). The cells were rinsed, lysed, and immunoprecipitated with 4 μg of rabbit antibodies to CrkL. The immunoprecipitates were separated on a 7.5% SDS-PAGE and analyzed by immunoblotting with the indicated antibodies.A, A short exposure of a blot probed with 4G10 to detect tyrosine-phosphorylated proteins. B, A longer exposure of A. EGF treatment rapidly changed the proteins associated with CrkL. NGF recruited an 84 kDa protein to CrkL.C, The CrkL immunoprecipitate was probed with antibodies to a number of signaling protein molecules. After EGF treatment a complex containing C3G/CrkL/Shp2/Cbl/EGFR was formed. In NGF-treated cells a complex was formed that contained C3G/CrkL/Shp2/Gab2/TrkA.D, CrkL was immunoprecipitated as in C, except that the cells were treated with NGF (50 ng/ml) for the indicated time intervals or the vehicle control (0 min). The immunoprecipitates were separated on SDS-PAGE, and the blot was probed with antibodies to Gab2, TrkA, and CrkL. In NGF-treated cells Gab2 and TrkA were both present in a complex with CrkL for the duration of the experiment. E, Cells were treated with NGF (50 ng/ml) for the indicated time intervals or the vehicle control (0 min), and TrkA was immunoprecipitated with MCTrk antibodies. The immunoprecipitates were separated on 10% SDS-PAGE, and the blot was probed with the following antibodies: pTrkA (pY490), total TrkA (MCTrk), and Gab2. All blots were visualized with SuperSignal; the results shown are representative of at least three independent experiments.
To identify the tyrosine-phosphorylated proteins that were associated either constitutively or transiently with CrkL, we probed the blots for a number of known signaling proteins (Fig. 4C). Our results revealed the following: (1) the 170 kDa tyrosine-phosphorylated protein that became associated with CrkL only after EGF treatment was the EGF receptor; (2) the 130 kDa constitutively tyrosine-phosphorylated protein was C3G; (3) the 120 kDa tyrosine-phosphorylated protein that transiently associated with CrkL in response to EGF was the adapter protein Cbl; (4) the tyrosine-phosphorylated 84 kDa protein that became associated with CrkL only in NGF-treated cells was the adapter protein Gab2 (Fig. 4C); and (5) the 68 kDa constitutively tyrosine-phosphorylated protein was the tyrosine phosphatase Shp2 (Fig.4C). Of note, the association of Gab2 with the CrkL-containing complex was quite persistent (Fig.4D). Although the maximal level of Gab2 was seen at 30 min after NGF treatment (Fig. 4D), a significant amount of Gab2 was still present in the CrkL immunocomplex at 60 min (Fig. 4D). When we reprobed the blots with a mouse anti-TrkA antibody, our results showed that TrkA also was recruited prominently to the CrkL complex at 2–5 min (Fig.4D). Although the amount of TrkA decreased at later times, it was still present in the complex at 60 min (Fig.4D). To examine whether NGF induced the association of Gab2 with TrkA, we immunoprecipitated TrkA from PC12 cells (Fig.4E). The blot was probed first with anti-pTrkA (pY490) antibodies, and the results showed that TrkA was activated in these cells even at 60 min after NGF treatment (Fig.4E). Although the total amount of TrkA did not appear to change, increasingly, Gab2 became associated with TrkA (Fig.4E).
Our finding showed that proteins known to regulate Rap1 activity are found in markedly different complexes in cells that have been treated with EGF and NGF. CrkL constitutively formed a complex with tyrosine-phosphorylated C3G and Shp2. This complex was changed rapidly by EGF treatment; C3G was dephosphorylated and a protein complex that included C3G/CrkL/Shp2/Cbl and tyrosine-phosphorylated EGFR was formed transiently. In contrast, C3G continued to be tyrosine-phosphorylated in NGF-treated cells, and the C3G/CrkL/Shp2 complex became stably associated with Gab2 and TrkA. These results raised the possibility that NGF treatment caused the formation of a complex in which activated TrkA is linked to activated Rap1.
We performed studies to define whether activated TrkA was associated with activated Rap1. Serum-starved PC12 cells were treated with NGF (50 ng/ml) for 30 min, a condition under which Rap1 is activated nearly maximally (Fig. 1C). Cells were rinsed and lysed in FB. The resulting supernatants from both control and NGF-treated samples were incubated with the RalGDSRBD/GST fusion protein to precipitate Rap1GTP as described above. The precipitates were analyzed by SDS-PAGE and immunoblotting with an antibody that specifically recognizes the activated form of TrkA (pY490). As shown in Figure 5, activated TrkA was detected only in the NGF-treated cells. Importantly, activated MAPK was also present in the precipitates of NGF-treated cells, but not control cells. We conclude that activated Rap1 is associated, either directly or indirectly, with activated TrkA and activated MAPK in NGF-treated cells. These data suggest that a signaling complex containing activated TrkA mediates the sustained activation of Rap1 and MAPK.
Fig. 5.

NGF treatment caused the association of activated TrkA with activated Rap1. Equal numbers of serum-starved PC12 cells were treated with NGF (50 ng/ml) or the vehicle control for 30 min. Rap1GTP was precipitated as described in Figure1C. The resulting precipitates were washed and separated by a 7.5% SDS-PAGE and transferred onto a PVDF membrane. The blot was probed with a rabbit antibody to either phosphorylated TrkA (pY490) or activated MAPK. All blots were visualized with SuperSignal; the results shown are representative of at least three independent experiments.
Evidence for the presence of Rap1 signaling complexes in early endosomes in NGF-treated cells
We next undertook studies to define the subcellular localization of activated Rap1. Several observations suggested that intracellular membranes, including endosomes, would be involved in Rap1 signaling. First, Rap1 has been shown to reside primarily in endosomes, secretory granules, and the Golgi apparatus (Kim et al., 1990; Beranger et al., 1991). Second, by 30 min of NGF treatment most surface TrkA receptors have been internalized (Grimes et al., 1996); this event coincides with the maximal activation of Rap1 seen in the current study. Importantly, in earlier studies examining crude endosomal fractions, NGF remained bound to TrkA (Beattie et al., 1996). We used confocal microscopy to investigate the subcellular localization of Rap1 and several components of the Rap1 signaling complex by determining whether or not they colocalized with EEA1, a marker for early endosomes (Mu et al., 1995).
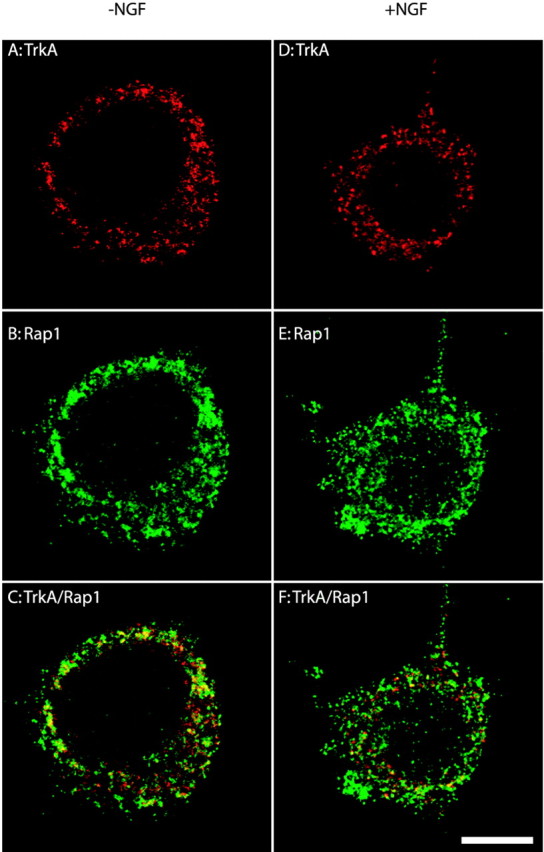
We first examined the distribution of TrkA, Rap1, and activated MAPK. PC12 cells were grown on Matrigel-coated coverslips. The cells were serum-starved for 12 hr. Then vehicle- and NGF-treated cells were processed for indirect immunofluorescence (Fig.6). We found that Rap1 was present in early endosomes in both control and NGF-treated PC12 cells. In vehicle-treated cells punctate EEA1 and Rap1 immunostaining decorated structures of various size that were distributed widely in the cytoplasm (Fig. 6C,D); partial colocalization of these markers was seen throughout the cytoplasm (Fig. 6C). After NGF treatment the immunostaining patterns for Rap1 and EEA1 were quite similar (Fig. 6D). There was increased colocalization of Rap1 and EEA1 with many bright puncta now clustered near the nucleus (Fig. 6D). Similar to previous observations (Beattie et al., 1996), punctate TrkA staining was distributed widely in the cytoplasm in vehicle-treated cells (Fig. 6A). There was little colocalization of TrkA and Rap1. After NGF treatment TrkA became concentrated in the perinuclear region and displayed significant colocalization with Rap1 (Fig. 6B). Next, we stained PC12 cells for activated MAPK. As expected, there was no signal for activated MAPK in vehicle-treated cells (Fig. 6E). However, in NGF-treated cells activated MAPK was seen in the perinuclear region where it was partially colocalized with EEA1 (Fig.6F). Taken together, these results are evidence that key components of the Rap1 signaling complex are present in endosomes.
Fig. 6.
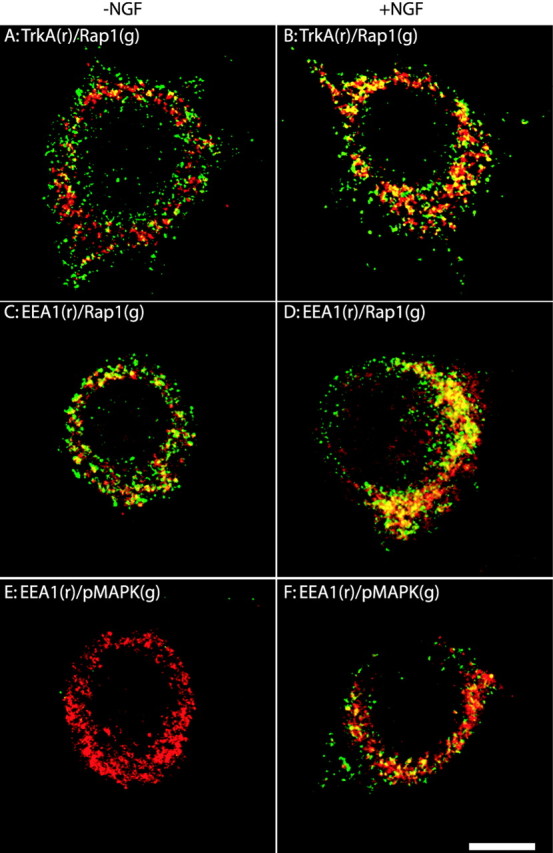
Analysis of the subcellular localization of components of the Rap1 signaling pathway by indirect immunofluorescence. PC12 cells were cultured for 24–48 hr on cover glasses that were coated with Matrigel. Cells were serum-starved and were treated with NGF (50 ng/ml; B, D, F) or the vehicle control (A, C, E) for 30 min. Then the cells were rinsed, fixed with 100% methanol, and processed for indirect immunofluorescence, as described in Materials and Methods. The primary antibodies were a rabbit antibody to Rap1 (1:250) and a mouse antibody to TrkA (1:400; A, B); a rabbit antibody to Rap1 (1:250) and a mouse antibody to EEA1 (1:120; C, D); a rabbit antibody to pMAPK (1:120) and a mouse antibody to EEA1 (1:120;E, F). Alexa 568 goat anti-mouse IgG conjugates and Alexa 488 goat anti-rabbit IgG conjugates were used to visualize the primary antibodies. g, Green fluorescence signal;r, red fluorescence signal. Colocalization of antigens is denoted by the yellow fluorescence signal. Scale bar, 10 μm.
Rap1 in intracellular membranes was activated by NGF
To define further the subcellular localization of Rap1, we used a step gradient density system of OptiPrep (5, 10, 15, 20, 25%) to fractionate PC12 cell homogenates (Fig.7A). Membrane fractions were collected and analyzed by SDS-PAGE and immunoblotting. An earlier study using similar gradients revealed that plasma membrane markers were present in low-density fractions and that internal membranes, such as early endosomes, were found in heavy-density fractions (Sheff et al., 1999). Consistent with these findings, we showed that plasma membrane, as marked by EGFR, was detected in fractions 1 and 2. The endosomal markers EEA1 and Rab5B were enriched in fractions 3 and 4 (Fig.7A).
Fig. 7.
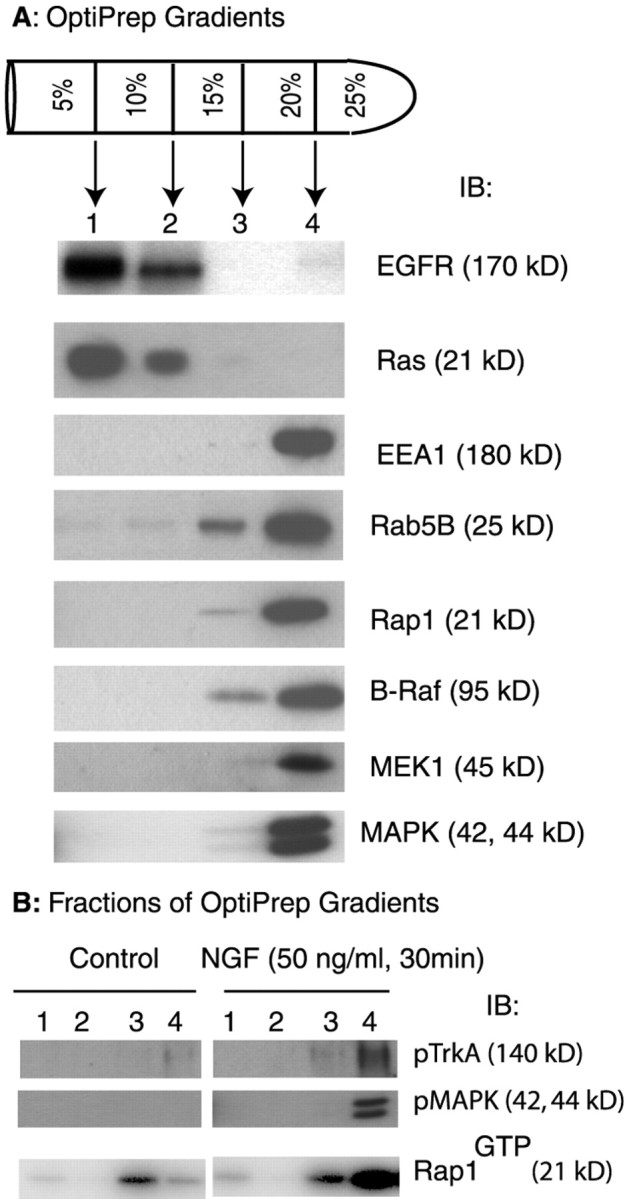
Activation of Rap1 in endosomal fractions.A, Postnuclear supernatants generated from equal numbers of serum-starved PC12 cells were fractionated by centrifugation, using a step gradient density system as described in Materials and Methods. Four membrane fractions were collected as indicated. Total proteins from each fraction were precipitated in 7% TCA and washed in acetone. Then the pellets were dried and boiled in SDS-PAGE loading buffer. The proteins were analyzed by SDS-PAGE and immunoblotting. The blot was probed independently with antibodies to EGFR, Ras, EEA1, Rab5B, Rap1, B-Raf, Mek1, and MAPK. The molecular mass corresponding to each antigen is shown in parentheses. B, Membrane fractions were collected from serum-starved PC12 cells that were treated with either NGF (50 ng/ml) or the vehicle control for 30 min. Total proteins from each fraction were precipitated, washed, and analyzed by SDS-PAGE and immunoblotting as in A. Activated TrkA and activated MAPK were detected by using specified antibodies. To detect activated Rap1, we collected and lysed membrane fractions in fishing buffer; the amount of Rap1GTPwas assayed as described in Figure 1. All blots were visualized with SuperSignal; the results shown are representative of at least three independent experiments.
There were striking differences for the fractionation of Ras and Rap1 in untreated cells. Although Ras was detected in lighter fractions (Fig. 7A), Rap1 was present only in heavier fractions. The presence of Ras in fractions 1 and 2 is consistent with its known plasma membrane localization (Leevers et al., 1994). Cofractionation of Rap1 with EEA1 and Rab5B is further evidence that Rap1 is localized in intracellular membranes (Fig. 7A). It is noteworthy that B-Raf, Mek1, and MAPK also were detected in fractions 3 and 4 (Fig. 7A) as were Gab2, C3G, and CrkL (data not shown).
To determine whether or not NGF induced Rap1 activation in endosome-enriched fractions, we treated serum-starved PC12 cells with either NGF (50 ng/ml) or the vehicle for 30 min. Step gradient density fractionation was performed as described above. The fractionation of Rap1 was unaffected by NGF treatment (data not shown). The membrane fractions collected from the gradient were adjusted to FB. Then the clear lysates were incubated with RalGDSRBD/GST to precipitate Rap1GTP. The precipitates were analyzed by SDS-PAGE and immunoblotting. A basal level of Rap1GTP was detected in the control sample (Fig. 7B). NGF treatment resulted in a marked increase in the amount of Rap1GTP in the heaviest fraction (Fig. 7B).
In a parallel experiment, proteins from each fraction were precipitated with TCA before analysis by SDS-PAGE and immunoblotting. We found that activated TrkA was detected only in membrane fractions isolated from NGF-treated cells (Fig. 7B). Significantly, activated TrkA was present only in the fractions enriched for Rap1. Similar results also were obtained for activated MAPK (Fig.7B). In preliminary studies we found that NGF treatment resulted in increased levels of Gab2, C3G, CrkL, B-Raf, Mek1, and MAPK in fraction 4 (data not shown). Our findings point to the existence of complexes containing activated TrkA together with activated Rap1 and activated MAPK in intracellular membranes in NGF-treated cells. Together with the results from immunostaining studies, our findings indicate that Rap1 signaling via the MAPK pathway can originate from endosomes.
BFA pretreatment prevented intracellular redistribution of TrkA and Rap1 in response to NGF
We envisioned that the activation of Rap1 would be sensitive to BFA. BFA has been shown to affect intracellular trafficking dramatically by disintegrating the Golgi apparatus and endosomes (Tooze and Hollinshead, 1992; Wood and Brown, 1992; Faundez et al., 1997), cellular compartments that contain Rap1 (Kim et al., 1990; Beranger et al., 1991). We first asked whether BFA treatment influenced the distribution of TrkA and Rap1. PC12 cells were grown and serum-starved as indicated in Figure 6. The cells were preincubated with BFA (5 μg/ml in 0.1% methanol) or the BFA vehicle (0.1% methanol) for 15 min at 37°C. Cells that either were treated with NGF (50 ng/ml, 30 min) or were left untreated then were processed for indirect immunofluorescence (Fig. 8). Preincubation with BFA did not appear to affect the amount of immunostaining or the distribution of TrkA or Rap1 (Fig.8A–C). However, in BFA-pretreated cells NGF treatment no longer resulted in the redistribution of TrkA and Rap1 to the perinuclear region (Fig. 8D,E), and there was little if any colocalization of TrkA and Rap1 (Fig.8F). These results are evidence that BFA disrupted the intracellular trafficking of TrkA and Rap1 after NGF treatment.
Fig. 8.
BFA pretreatment disrupted the intracellular trafficking of TrkA and Rap1. PC12 cells were cultured for 24–48 hr on cover glasses coated with Matrigel. Cells were serum-starved and were pretreated with 5 μg/ml BFA at 37°C for 15 min. Then the cells were treated with 50 ng/ml NGF (D, E, F) or the vehicle control (A, B, C) for 30 min. Cells were rinsed, fixed with 100% methanol, and processed for indirect immunofluorescence, as described in Materials and Methods. Immunostaining was performed with a mouse antibody to TrkA (1:400;A, D) and a rabbit antibody to Rap1 (1:250; B, E). Alexa 568 goat anti-mouse IgG conjugates and Alexa 488 goat anti-rabbit IgG conjugates were used to visualize TrkA and Rap1, respectively. C, A merged image of A andB. F, A merged image of Dand E. Pretreatment with the BFA vehicle control (0.1% methanol) alone had no effect on the intracellular trafficking of TrkA and Rap1 after NGF treatment (data not shown). Colocalization of antigens is denoted by the yellow fluorescence signal. Scale bar, 10 μm.
BFA inhibited Rap1 activation and the sustained activation of MAPK by NGF
Given the disruptive effect of BFA on the intracellular trafficking of TrkA and Rap1, we reasoned that BFA pretreatment would affect NGF-induced Rap1 signaling in PC12 cells. To determine whether BFA significantly changed signaling events at the plasma membrane, we preincubated the cells with either BFA (5 μg/ml in 0.1% methanol) or the vehicle (0.1% methanol) and then treated them with NGF (50 ng/ml) as indicated (Fig. 9). BFA pretreatment had no significant effect on TrkA activation (data not shown) or on the extent or the time course of Ras activation (Fig. 9A), nor was there a change in the total amount of Ras (Fig. 9A). In contrast, BFA pretreatment significantly inhibited NGF-induced Rap1 activation (Fig. 9B). The inhibitory effect was seen at early time points (2, 5 min) but was more pronounced at later time points (30, 60 min) when maximal Rap1 activation was present in cells not pretreated with BFA (Fig. 9B). We conclude that BFA selectively blocked Rap1 activation in NGF-treated cells.
Fig. 9.

Inhibition of NGF-induced Rap1 activation by BFA pretreatment. Equal numbers of serum-starved PC12 cells were pretreated with either 5 μg/ml BFA (+) or the vehicle control (−) at 37°C for 30 min. Then the cells were treated with NGF (50 ng/ml) for the indicated time intervals or the vehicle control (0 min). Activated Ras (A) and Rap1 (B) were assayed as described in Figure 1. C, PC12 cells were pretreated with either 5 μg/ml BFA (+) or the vehicle control (−) at 37°C for 15 min. Then the cells were treated with 50 ng/ml NGF for 5 or 30 min or the vehicle control (0 min). Cells were rinsed and lysed in RIPA buffer; cell lysate proteins (20 μg) were separated on 10% SDS-PAGE, transferred, and blotted with specific antibodies against activated MAPK and total MAPK. All blots were visualized with SuperSignal; the results shown are representative of at least three independent experiments.
In view of our findings for the existence of complexes containing activated Rap1 and MAPK, we predicted that BFA pretreatment also would prevent the persistent activation of MAPK. To show whether or not this was the case, we analyzed MAPK in BFA-pretreated cells. As shown in Figure 9C, BFA pretreatment had little effect on NGF-induced activation of MAPK at 5 min but abolished the persistent activation of MAPK at 30 min (Fig. 9C, top). These observations suggest that a BFA-sensitive subcellular compartment is required for Rap1 activation and for the persistent activation of MAPK induced by NGF.
DISCUSSION
The unique architecture of neurons imposes the need to move neurotrophic signals over long distances. The molecular mechanisms that serve this need are yet to be elucidated. One hypothesis is that signaling endosomes carry the neurotrophic signal (Grimes et al., 1996,1997). If true, signaling endosomes must ensure the propagation of signals that are specific and long-lived. We demonstrated that NGF acted via TrkA to induce the persistent activation of endogenous Rap1 and prolonged activation of the MAPK pathway as well as the formation of a signaling complex in which activated Rap1 was present together with activated TrkA and activated MAPK. In studies to localize Rap1 signaling, we showed that activated Rap1 was present with the other elements of the signaling complex in early endosomes. By disrupting the endosomal compartment with BFA pretreatment, we selectively inhibited NGF-induced Rap1 activation and persistent MAPK activation. Rap1 signaling in endosomes thus appears to contribute to long-lived NGF signaling via the MAPK pathway.
A role for Rap1 in the NGF-induced sustained activation of MAPK
Although both Ras and Rap1 have been implicated in NGF-induced MAPK activation in previous studies (Bar-Sagi and Feramisco, 1985;Hagag et al., 1986; Qui and Green, 1991, 1992; York et al., 1998), whether NGF induces the activation of endogenous Rap1 to activate the MAPK pathway has been controversial (York et al., 1998; Zwartkruis et al., 1998). To examine the contributions of Ras and Rap1 to NGF signaling, we investigated the activation of endogenous Ras and Rap1 in PC12 cells. After confirming that sustained MAPK activation was induced by NGF, but not by EGF, we examined the temporal patterns of Ras activation. We showed that Ras was activated transiently by both NGF and EGF. We conclude that it is unlikely that activation of endogenous Ras accounts for the prolonged activation of MAPK seen in NGF-treated cells. Our findings differ from those in earlier studies in which oncogenic Ras was introduced into PC12 cells (Bar-Sagi and Feramisco, 1985; Qui and Green, 1992). Although persistently activated oncogenic Ras led to prolonged activation of MAPK in these studies, our results reveal that under normal circumstances Ras-dependent signaling is activated only transiently. Consistent with our observations, York et al. (1998) demonstrated that a dominant-negative Ras mutant inhibited the early phase, but not the late phase, of MAPK activation in PC12 cells after NGF treatment. Therefore, a factor or factors other than Ras must contribute to the prolonged activation of MAPK induced by NGF. Our findings do not obviate a role for Ras signaling in the differentiation of PC12 cells. Indeed, the injection of anti-Ras antibodies or the expression of a dominant-negative Ras mutant inhibited neurite outgrowth in response to NGF (Hagag et al., 1986;Vossler et al., 1997). In concert with others (York et al., 1998), we suggest that multiple signaling pathways contribute to the differentiation of PC12 cells.
In the present study we show that the activation of endogenous Rap1 correlates temporally with the prolonged activation of MAPK induced by factors such as NGF and bFGF, growth factors that cause differentiation in PC12 cells. Although the exact role of Rap1 in neuronal function remains to be elucidated, an increasing body of evidence suggests that Rap1 plays an important role in neuronal signaling leading to differentiation. We have shown that the inhibition of Rap1 activation by pretreatment with BFA selectively abolished the persistent activation of MAPK in NGF-treated cells. Using a different approach,York et al. (2000) demonstrated that the inhibition of Rap1 activity by overexpressing Rap1GAP1 selectively blocked the sustained phase of MAPK activation induced by NGF. Finally, Lu et al. (2000) reported that when RalGDS-RBD was expressed to block Rap1 signaling, NGF-dependent neurite outgrowth was inhibited in PC12-Shb cells. These results point to a significant role played by Rap1 in neuronal signaling. Additional studies are warranted to pursue a role for activated endogenous Rap1 in inducing neuronal differentiation.
Regulation of Rap1 activation: NGF induces a novel complex containing C3G and CrkL
The sustained Rap1 response that followed NGF treatment matched the temporal pattern of MAPK activation, suggesting that the two responses are linked. To explore this, we compared the signaling proteins in complex with C3G and CrkL in NGF- and EGF-treated cells. Tyrosine phosphorylation of C3G has been shown to be required for Rap1 activation in COS cells (Ichiba et al., 1999). Depending on the cell type, C3G regulates Rap1 signaling via either CrkII (Okada and Pessin, 1997) or CrkL (Boussiotis et al., 1997). In previous studies maximal activation of Rap1/B-Raf signaling in PC12 cells was seen when C3G was cotransfected with CrkL, but not with CrkII (York et al., 1998).
There were marked differences in the complexes that contained C3G and CrkL after treatment with EGF or NGF. In untreated cells C3G was tyrosine phosphorylated constitutively and formed a stable complex with CrkL and Shp2. EGF rapidly induced marked dephosphorylation of C3G and rapid recruitment of the C3G/CrkL/Shp2 complex to EGFR, possibly via the adapter Cbl. Because Cbl has been shown to regulate receptor and nonreceptor tyrosine kinase signaling pathways negatively (Ota and Samelson, 1997), it is possible that the association of C3G/CrkL/Shp2 with Cbl interferes with the ability of C3G to activate Rap1. In additional immunoprecipitation experiments with antibodies to Cbl and Shp2, we confirmed the assignment of C3G, CrkL, Cbl, Shp2, and EGFR to the complexes found in EGF-treated cells (data not shown).
In contrast to the findings with EGF, C3G was tyrosine phosphorylated clearly and persistently after NGF treatment. NGF resulted in the binding of C3G/CrkL/Shp2 to Gab2 and TrkA. This complex was long-lived, persisting for at least 60 min. It is tempting to suggest that Gab2 is an adapter that links TrkA to the complex. Gab2 is a member of the insulin receptor substrate family and plays an important role in cytokine- and growth factor receptor-mediated signaling (Nishida et al., 1999; Zhao et al., 1999). In fibroblasts, Gab2 was found to suppress Ras-dependent signaling (Zhao et al., 1999), an effect that is consistent with a role for Gab2 in activating Rap1 signaling. Sequence analysis has revealed that Gab2 contains a pleckstrin homology motif by which it could bind directly to receptor tyrosine kinases such as TrkA. Gab2 also possesses CrkL-, PI3K-, and Shp2-binding domains (Holgado-Madruga et al., 1997; Korhonen et al., 1999; Nishida et al., 1999). Whether Gab2 directly or indirectly binds to TrkA warrants further investigation. Gab1, a Gab2 homolog, recently has been reported to modulate the TrkA signaling leading to differentiation (Korhonen et al., 1999). Using commercially available antibodies, we were unable to detect Gab1 in CrkL immunoprecipitations in our studies, nor were we able to detect FRS-2 in the CrkL-containing complex as reported by Kao et al. (2001).
The existence of the C3G/CrkL/Shp2/Gab2/TrkA complex in NGF-treated cells suggests that activated TrkA might be present in a complex with activated Rap1. In NGF-treated cells we found that activated TrkA was coprecipitated specifically with activated Rap1. Importantly, such precipitates also contained activated MAPK. Although further studies are needed to define these complexes, the results suggest that activated Rap1 is linked, directly or indirectly, to the activated forms of both TrkA and MAPK and that this complex contributes to NGF-induced persistent MAPK activation.
Rap1 signaling from endosomes
A number of observations suggest that Rap1 signaling arises from endosomes. First, Rap1 is localized principally in intracellular membranes. Second, most TrkA receptors at the cell surface have been internalized by 30 min of treatment (Grimes et al., 1996). Third, York and colleagues (2000) showed that internalization of TrkA after NGF treatment is required for Rap1 activation in PC12 cells. Fourth, after NGF treatment one can isolate endosomes that contain NGF bound to TrkA, and activated TrkA is present in endosomes at 30 min (Beattie et al., 1996). Finally, we consistently detected a 30 min delay in maximal Rap1 activation, suggesting that events downstream from TrkA activation are required for the full activation of Rap1. We examined NGF effects on the localization of Rap1 and the components of the Rap1 signaling complex by confocal microscopy. NGF treatment consistently resulted in the presence of TrkA, Rap1, B-Raf, and activated MAPK in the perinuclear region. Importantly, we found that TrkA colocalized with Rap1 and that Rap1, B-Raf, and activated MAPK colocalized with EEA1 near the nucleus after NGF treatment. These data are evidence that the Rap1 signaling complex resides in early endosomes.
We used cellular fractionation to define further the cellular compartment or compartments that contained TrkA and Rap1. In both treated and untreated cells Rap1 was present in intracellular membranes that cofractionated with the endosomal markers EEA1 and Rab5B. The levels of B-Raf, Mek1, and MAPK were increased by NGF treatment in these fractions, as were the levels of Gab2, CrkL, and C3G. Significantly, these membranes also contained activated TrkA and activated MAPK. Our results suggest that after NGF treatment active Rap1 signaling complexes are formed on endosomal membranes to convey the NGF signal to the MAPK pathway. In preliminary studies we have shown that these endosomal fractions are indeed competent to convey the signal to Elk-1 in an in vitro kinase assay (data not shown).
Disrupting intracellular membranes inhibited signaling via Rap1
Because Rap1 is concentrated in the Golgi apparatus and endosomes, we reasoned that disrupting these compartments with BFA would inhibit Rap1 activation. Pretreatment with BFA markedly changed the trafficking of TrkA and Rap1 in NGF-treated cells. NGF treatment no longer caused TrkA and Rap1 to be concentrated and colocalized in the perinuclear region. Significantly, BFA pretreatment also inhibited Rap1 activation and the prolonged activation of MAPK. These effects of BFA appeared to be specific, because BFA pretreatment showed no detectable effect on Ras activation or on the early phase of MAPK activation. Thus the integrity of a BFA-sensitive intracellular compartment or compartments is critical for the activation of Rap1 signaling induced by NGF.
Different subcellular compartments for Ras and Rap1 signaling
Our present studies suggest that NGF signaling to MAPK uses at least two pathways. After the activation of TrkA by NGF, Ras-dependent signaling is activated immediately from caveolae in the plasma membrane (Wu et al., 1997; Anderson, 1998; Huang et al., 1999). In addition to its effects on activation of the C-Raf/Mek1/MAPK pathway at the plasma membrane, we have demonstrated that Ras together with activated MAPK is carried on clathrin-coated vesicles (CCVs) into NGF-treated cells. CCVs appear to function as one class of signaling endosomes (C. L. Howe, E. C. Beattie, J. C. Valletta, and W. C. Mobley, unpublished observation).
The second pathway, one that appears to distinguish NGF and EGF signaling, is initiated when activated TrkA associates with C3G/CrkL/Shp2/Gab2. Where this complex is formed is unclear. However, our findings suggest that it is trafficked with TrkA to the endosome where C3G interacts with Rap1, thus triggering sustained Rap1 activation and prolonged B-Raf/Mek1/MAPK signaling. Prolonged Rap1 activation in endosomes may allow the NGF signal to be moved from axon terminals to neuronal cell bodies where effects on cellular differentiation are elicited. We speculate that signaling endosomes containing activated Rap1 may be a platform from which diverse signaling events can be initiated robustly, persistently, and specifically.
Footnotes
These studies were funded by National Institutes of Health Grant NS24054, the McGowan Charitable Fund, and a gift from the Powell Foundation. We are grateful to the members of the Mobley lab for their invaluable input. We also thank Drs. Pingsheng Liu, Richard G. W. Anderson, Phil Stork, and Randall York for their constructive comments.
Correspondence should be addressed to Dr. Chengbiao Wu, Program in Neuroscience, Stanford University School of Medicine, Medical School Laboratory Surge Building, Room P222, 1201 Welch Road, Stanford, CA 94305. E-mail: cbwu@stanford.edu.
REFERENCES
- 1.Anderson RGW. The caveolae membrane system. Annu Rev Biochem. 1998;67:199–225. doi: 10.1146/annurev.biochem.67.1.199. [DOI] [PubMed] [Google Scholar]
- 2.Bar-Sagi D, Feramisco JR. Microinjection of the ras oncogene protein into PC12 cells induces morphological differentiation. Cell. 1985;42:841–848. doi: 10.1016/0092-8674(85)90280-6. [DOI] [PubMed] [Google Scholar]
- 3.Beattie EC, Zhou J, Grimes ML, Bunnett NW, Howe CL, Mobley WC. A signaling endosome hypothesis to explain NGF actions: potential implications for neurodegeneration. Cold Spring Harb Symp Quant Biol. 1996;61:389–406. [PubMed] [Google Scholar]
- 4.Beranger F, Goud B, Tavitian A, de Gunzburg J. Association of the Ras-antagonistic Rap1/Krev-1 proteins with the Golgi complex. Proc Natl Acad Sci USA. 1991;88:1606–1610. doi: 10.1073/pnas.88.5.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bos JL. All in the family? New insights and questions regarding interconnectivity of Ras, Rap1, and Ral. EMBO J. 1998;17:6776–6782. doi: 10.1093/emboj/17.23.6776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boulton TG, Nye SH, Robbins DJ, Ip NY, Radziejewska E, Morgenbesser SD, DePinho RA, Panayotatos N, Cobb MH, Yancopoulos GD. ERKs: a family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell. 1991;65:663–675. doi: 10.1016/0092-8674(91)90098-j. [DOI] [PubMed] [Google Scholar]
- 7.Boussiotis VA, Freeman GI, Berezovskaya A, Barber DL, Nadler LM. Maintenance of human T-cell anergy: blocking of IL-2 gene transcription by activated Rap1. Science. 1997;278:124–128. doi: 10.1126/science.278.5335.124. [DOI] [PubMed] [Google Scholar]
- 8.Chao M, Casaccia-Bonnefil P, Carter B, Chittka A, Kong H, Yoon SO. Neurotrophin receptors: mediators of life and death. Brain Res Brain Res Rev. 1998;26:295–301. doi: 10.1016/s0165-0173(97)00036-2. [DOI] [PubMed] [Google Scholar]
- 9.Cowley S, Paterson H, Kemp P, Marshall CJ. Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell. 1994;77:841–852. doi: 10.1016/0092-8674(94)90133-3. [DOI] [PubMed] [Google Scholar]
- 10.Ehlers MD, Kaplan DR, Price DL, Koliatsos VE. NGF-stimulated retrograde transport of trkA in the mammalian nervous system. J Cell Biol. 1995;130:149–156. doi: 10.1083/jcb.130.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Faundez V, Horng J-T, Kelly RB. ADP ribosylation factor 1 is required for synaptic vesicle budding in PC12 cells. J Cell Biol. 1997;138:505–515. doi: 10.1083/jcb.138.3.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gotoh T, Hattori S, Nakamura S, Kitayama H, Noda M, Takai Y, Kaibuchi K, Matui H, Hatase O, Takahashi H, Kurata T, Matsuda M. Identification of Rap1 as a target for the Crk SH3 domain-binding guanine nucleotide-releasing factor C3G. Mol Cell Biol. 1995;15:6746–6753. doi: 10.1128/mcb.15.12.6746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greene LA, Tischler AS. PC12 pheochromocytoma culture in neurobiological research. Adv Cell Neurobiol. 1982;3:373–414. [Google Scholar]
- 14.Grewal SS, York RD, Stork PJ. Extracellular signal-regulated kinase signaling in neurons. Curr Opin Neurobiol. 1999;9:544–553. doi: 10.1016/S0959-4388(99)00010-0. [DOI] [PubMed] [Google Scholar]
- 15.Grimes ML, Zhou J, Beattie EC, Yuen EC, Hall DE, Valletta JS, Topp KS, LaVail JH, Bunnett NW, Mobley WC. Endocytosis of activated TrkA: evidence that nerve growth factor induces formation of signaling endosomes. J Neurosci. 1996;16:7950–7964. doi: 10.1523/JNEUROSCI.16-24-07950.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grimes ML, Beattie EC, Mobley WC. A signaling organelle containing the nerve growth factor-activated receptor tyrosine kinase, TrkA. Proc Natl Acad Sci USA. 1997;94:9909–9914. doi: 10.1073/pnas.94.18.9909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hagag N, Halegoua S, Viola M. Inhibition of growth factor-induced differentiation of PC12 cells by microinjection of antibody to Ras p21. Nature. 1986;319:680–682. doi: 10.1038/319680a0. [DOI] [PubMed] [Google Scholar]
- 18.Herrmann C, Martin GA, Wittinghofer A. Quantitative analysis of the complex between p21ras and the Ras-binding domain of the human Raf-1 protein kinase. J Biol Chem. 1995;270:2901–2905. doi: 10.1074/jbc.270.7.2901. [DOI] [PubMed] [Google Scholar]
- 19.Herrmann C, Horn G, Spaargaren M, Wittinghofer A. Differential interaction of the Ras family GTP-binding proteins H-Ras, Rap1A, and R-Ras with the putative effector molecules Raf kinase and Ral-guanine nucleotide exchange factor. J Biol Chem. 1996;271:6794–6800. doi: 10.1074/jbc.271.12.6794. [DOI] [PubMed] [Google Scholar]
- 20.Holgado-Madruga M, Moscatello DK, Emlet DR, Dieterich R, Wong AJ. Grb2-associated binder-1 mediates phosphatidyl inositol 3-kinase activation and the promotion of cell survival by nerve growth factor. Proc Natl Acad Sci USA. 1997;94:12419–12424. doi: 10.1073/pnas.94.23.12419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Howe CL, Mobley WC. Nerve growth factor effects on cholinergic modulation of hippocampal and cortical plasticity. In: Mocchettic I, editor. Neurobiology of the neurotrophins. Graham Publishing; Johnson City, TN: 2001. pp. 255–307. [Google Scholar]
- 22.Huang CS, Zhou J, Feng AK, Lynch CC, Klumperman J, DeArmond SJ, Mobley WC. Nerve growth factor signaling in caveolae-like domains at the plasma membrane. J Biol Chem. 1999;274:36707–36714. doi: 10.1074/jbc.274.51.36707. [DOI] [PubMed] [Google Scholar]
- 23.Ichiba T, Hashimoto Y, Nakaya M, Kuraishi Y, Tanaka S, Kurata T, Mochizuki N, Matsuda M. Activation of C3G guanine nucleotide exchange factor for Rap1 by phosphorylation of tyrosine 504. J Biol Chem. 1999;274:14376–14381. doi: 10.1074/jbc.274.20.14376. [DOI] [PubMed] [Google Scholar]
- 24.Kao S, Jaiswal RK, Kolch W, Landreth GE. Identification of the mechanisms regulating the differential activation of the MAP kinase cascade by epidermal growth factor and nerve growth factor in PC12 cells. J Biol Chem. 2001;276:18169–18177. doi: 10.1074/jbc.M008870200. [DOI] [PubMed] [Google Scholar]
- 25.Kim S, Mizoguchi A, Kikuchi A, Takai Y. Tissue and subcellular distributions of the smg-21/Rap1/Krev-1 proteins which are partly distinct from those of c-Ras p21s. Mol Cell Biol. 1990;10:2645–2652. doi: 10.1128/mcb.10.6.2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Korhonen JM, Said FA, Wong AJ, Kaplan DR. Gab1 mediates neurite outgrowth, DNA synthesis, and survival in PC12 cells. J Biol Chem. 1999;274:37307–37314. doi: 10.1074/jbc.274.52.37307. [DOI] [PubMed] [Google Scholar]
- 27.Leevers SJ, Paterson HF, Marshall CJ. Requirement for Ras in Raf activation is overcome by targeting Raf to the plasma membrane. Nature. 1994;369:411–414. doi: 10.1038/369411a0. [DOI] [PubMed] [Google Scholar]
- 28.Lu L, Anneren C, Reedquist KA, Bos JL, Welsh M. NGF-dependent neurite outgrowth in PC12 cells overexpressing the Src homology 2-domain protein shb requires activation of the Rap1 pathway. Exp Cell Res. 2000;259:370–377. doi: 10.1006/excr.2000.4984. [DOI] [PubMed] [Google Scholar]
- 29.Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- 30.Meakin SO, Shooter EM. The nerve growth factor family of receptors. Trends Neurosci. 1992;15:323–331. doi: 10.1016/0166-2236(92)90047-c. [DOI] [PubMed] [Google Scholar]
- 31.Mobley WC, Rutkowski JL, Tennekoon GI, Gemski J, Buchanan K, Johnston MV. Nerve growth factor increases choline acetyltransferase activity in developing basal forebrain neurons. Brain Res. 1986;387:53–62. doi: 10.1016/0169-328x(86)90020-3. [DOI] [PubMed] [Google Scholar]
- 32.Mu FT, Callaghan JM, Steele-Mortimer O, Stenmark H, Parton RG, Campbell PL, McCluskey J, Yeo JP, Tock EP, Toh BH. EEA1, an early endosome-associated protein: EEA1 is a conserved α-helical peripheral membrane protein flanked by cysteine “fingers” and contains a calmodulin-binding IQ motif. J Biol Chem. 1995;270:13503–13511. doi: 10.1074/jbc.270.22.13503. [DOI] [PubMed] [Google Scholar]
- 33.Nishida K, Yoshida Y, Itoh M, Fukada T, Ohtani T, Shirogane T, Atsumi T, Takahashi-Tezuka M, Ishihara K, Hibi M, Hirano T. Gab-family adapter proteins act downstream of cytokine and growth factor receptors and T- and B-cell antigen receptors. Blood. 1999;93:1809–1816. [PubMed] [Google Scholar]
- 34.Okada S, Pessin JE. Insulin and epidermal growth factor stimulate a conformational change in Rap1 and dissociation of the CrkII–C3G complex. J Biol Chem. 1997;272:28179–28182. doi: 10.1074/jbc.272.45.28179. [DOI] [PubMed] [Google Scholar]
- 35.Ota Y, Samelson LE. The product of the proto-oncogene c-cbl: a negative regulator of the Syk tyrosine kinase. Science. 1997;276:418–420. doi: 10.1126/science.276.5311.418. [DOI] [PubMed] [Google Scholar]
- 36.Pang L, Sawada T, Decker SJ, Saltiel AR. Inhibition of MAP kinase kinase blocks the differentiation of PC-12 cells induced by nerve growth factor. J Biol Chem. 1995;270:13585–13588. doi: 10.1074/jbc.270.23.13585. [DOI] [PubMed] [Google Scholar]
- 37.Qui MS, Green SH. NGF and EGF rapidly activate p21ras in PC12 cells by distinct, convergent pathways involving tyrosine phosphorylation. Neuron. 1991;7:937–946. doi: 10.1016/0896-6273(91)90339-2. [DOI] [PubMed] [Google Scholar]
- 38.Qui MS, Green SH. PC12 cell neuronal differentiation is associated with prolonged p21ras activity and consequent prolonged ERK activity. Neuron. 1992;9:705–717. doi: 10.1016/0896-6273(92)90033-a. [DOI] [PubMed] [Google Scholar]
- 39.Riccio A, Pierchala BA, Ciarallo CL, Ginty DD. An NGF-TrkA-mediated retrograde signal to transcription factor CREB in sympathetic neurons. Science. 1997;277:1097–1100. doi: 10.1126/science.277.5329.1097. [DOI] [PubMed] [Google Scholar]
- 40.Riccio A, Ahn S, Davenport CM, Blendy JA, Ginty DD. Mediation by a CREB family transcription factor of NGF-dependent survival of sympathetic neurons. Science. 1999;286:2358–2361. doi: 10.1126/science.286.5448.2358. [DOI] [PubMed] [Google Scholar]
- 41.Sandow S, Heydon K, Weible M, Reynolds A, Bartlett S, Hendry I. Signaling organelle for retrograde axonal transport of internalized neurotrophins from the nerve terminal. Immunol Cell Biol. 2000;78:430–435. doi: 10.1046/j.1440-1711.2000.00924.x. [DOI] [PubMed] [Google Scholar]
- 42.Sheff DR, Daro EA, Hull M, Mellman I. The receptor recycling pathway contains two distinct populations of early endosomes with different sorting functions. J Cell Biol. 1999;145:123–139. doi: 10.1083/jcb.145.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stephens RM, Loeb DM, Copeland T, Pawson T, Greene LA, Kaplan DR. Trk receptors use redundant signal transduction pathways involving Shc and PLC-γ1 to mediate NGF response. Neuron. 1994;12:691–705. doi: 10.1016/0896-6273(94)90223-2. [DOI] [PubMed] [Google Scholar]
- 44.Tooze J, Hollinshead M. In AtT20 and HeLa cells brefeldin A induces the fusion of tubular endosomes and changes their distribution and some of their endocytic properties. J Cell Biol. 1992;118:813–830. doi: 10.1083/jcb.118.4.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vossler MR, Yao H, York RD, Pan MG, Rim CS, Stork PJ. cAMP activates MAP kinase and Elk-1 through a B-Raf- and Rap1-dependent pathway. Cell. 1997;89:73–82. doi: 10.1016/s0092-8674(00)80184-1. [DOI] [PubMed] [Google Scholar]
- 46.Wood SA, Brown WJ. The morphology but not the function of endosomes and lysosomes is altered by brefeldin A. J Cell Biol. 1992;119:273–285. doi: 10.1083/jcb.119.2.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu C, Butz S, Ying Y-s, Anderson RGW. Tyrosine kinase receptors concentrated in caveolae-like domains from neuronal plasma membrane. J Biol Chem. 1997;272:3554–3559. doi: 10.1074/jbc.272.6.3554. [DOI] [PubMed] [Google Scholar]
- 48.York RD, Yao H, Dillon T, Ellig CL, Eckert SP, McCleskey EW, Stork PJ. Rap1 mediates sustained MAP kinase activation induced by nerve growth factor. Nature. 1998;392:622–626. doi: 10.1038/33451. [DOI] [PubMed] [Google Scholar]
- 49.York RD, Molliver DC, Grewal SS, Stenberg PE, McCleskey EW, Stork PJ. Role of phosphoinositide 3-kinase and endocytosis in nerve growth factor-induced extracellular signal-regulated kinase activation via Ras and Rap1. Mol Cell Biol. 2000;21:8069–8083. doi: 10.1128/mcb.20.21.8069-8083.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Young SW, Dickens M, Tavare JM. Differentiation of PC12 cells in response to a cAMP analogue is accompanied by sustained activation of mitogen-activated protein kinase: comparison with the effects of insulin, growth factors, and phorbol esters. FEBS Lett. 1994;338:212–216. doi: 10.1016/0014-5793(94)80367-6. [DOI] [PubMed] [Google Scholar]
- 51.Zhao C, Yu DH, Shen R, Feng GS. Gab2, a new pleckstrin homology domain-containing adapter protein, acts to uncouple signaling from ERK kinase to Elk-1. J Biol Chem. 1999;274:19649–19654. doi: 10.1074/jbc.274.28.19649. [DOI] [PubMed] [Google Scholar]
- 52.Zwartkruis FJ, Wolthuis RM, Nabben NM, Franke B, Bos JL. Extracellular signal-regulated activation of Rap1 fails to interfere in Ras effector signaling. EMBO J. 1998;17:5905–5912. doi: 10.1093/emboj/17.20.5905. [DOI] [PMC free article] [PubMed] [Google Scholar]