

Abstract
Background
Since miltefosine monotherapy against visceral leishmaniasis (VL) caused by Leishmania donovani has been discontinued in the Indian subcontinent due to an increase in the number of treatment failures, single dose liposomal amphotericin B is now advocated as a treatment option of choice. Paromomycin-miltefosine combination therapy can be used as substitute first-line treatment in regions without cold-chain potential. Previous laboratory studies in the closely related species Leishmania infantum have demonstrated that paromomycin monotherapy fairly rapidly selects for resistance producing a phenotype with increased fitness. Given the possible clinical implications of these findings for the current field situation, the present study aimed to identify the potential hazards of paromomycin-miltefosine combination therapy.
Principal findings
Drug interaction studies using the fixed-ratio isobologram method revealed an indifferent interaction between paromomycin and miltefosine. In hamsters infected with L. infantum, the combination resulted in cumulative efficacy in reducing parasite burdens in the liver, spleen and bone marrow. Selected resistant lines against the single drugs did not display cross-resistance. When the intracellular amastigote stage was repeatedly exposed to the paromomycin-miltefosine combination, either in vitro or in vivo, no significant susceptibility decrease towards either drug was noted.
Conclusions
These results suggest that implementation of paromomycin-miltefosine combination therapy indeed could represent a safe and affordable treatment option for L. donovani VL as miltefosine appears to overrule the anticipated rapid development of PMM resistance.
Author summary
Liposomal amphotericin B is presently being used as first-line treatment option against visceral leishmaniasis in the Indian subcontinent. However, the need for temperature-controlled transport and storage limits its widespread use in rural areas. Previous studies already suggested that paromomycin-miltefosine combination therapy could be a valuable alternative, side passing some of the disadvantages associated with monotherapy, such as development of drug resistance. As the first reports of miltefosine resistant clinical isolates have already surfaced and paromomycin resistance could be easily induced under laboratory conditions, it remains essential to assess the risk of developing resistance against both drugs upon combination therapy. This study evaluated the efficacy of combined therapy against a Leishmania species closely related to the agent found in the Indian subcontinent, using both in vitro and in vivo models with the aim to select multidrug-resistant species by simultaneous exposure to paromomycin and miltefosine. The combination of both drugs in the hamster model resulted in a cumulative efficacy but did not lead to a significant susceptibility decrease, indicating that paromomycin-miltefosine combination therapy may represent a safe and affordable treatment option for visceral leishmaniasis.
Introduction
Depending on the geographical location, visceral leishmaniasis (VL) can either be caused by Leishmania donovani (East Africa and Indian subcontinent) or L. infantum (Mediterranean basin, central Asia and Latin America) [1]. Although both species are closely related and belong to the same complex (L. donovani complex), transmission of L. donovani is mostly anthroponotic while the domestic dog serves as the main reservoir of L. infantum. Various treatment recommendations for VL have been proposed in the past and up to the early 2000’s most cases were treated with antimonials (Sb) [2]. However, the spread of Sb-resistant parasites in the Indian subcontinent has enforced a shift in therapeutic modalities [3]. Given its limited toxic effects, oral administration and reasonable price, the introduction of miltefosine (MIL) in 2002 as a novel antileishmanial agent looked very promising. As a result, the drug was even presented as first-line therapy in 2005 in India, Bangladesh and Nepal in the frame of the Kala-azar elimination program aimed at reducing the disease burden to less than 1/1000 by 2015 [4]. Unfortunately, recent studies in the Indian subcontinent demonstrated an increasing number of MIL-treatment failures hence limiting its further use in monotherapy [5] and leading to the recommendation of a single dose of liposomal amphotericin B (L-AmB) as treatment option of choice [6, 7]. However, the requirement for temperature-controlled cold-chain facilities to transport and preserve L-AmB restricts its widespread use. In regions without cold-chain assurance, the short-term combination of MIL and paromomycin (PMM) has been suggested as alternative [8]. The development of MIL-resistance in monotherapy can be anticipated as a major limitation given its long elimination half-life and long treatment course [9]. Although no decreased MIL-susceptibility could yet be demonstrated in vitro in isolates of L. donovani derived from treatment failures in the Indian subcontinent, the first actual MIL-resistant clinical Leishmania isolates did already surface [10–12]. Rapid development of laboratory-induced PMM-resistance has been demonstrated [11, 13–15] which also proved to be associated with a potential fitness gain [16, 17] paving a way to rapid emergence and spread of PMM-resistance upon its routine use in the field. Although drug resistance would theoretically arise slower for drug combinations, it remains essential to assess the potential effect of introducing PMM-MIL combination therapy on a large scale, given the above mentioned limitations of PMM- or MIL-monotherapy. This laboratory study assessed the interaction between MIL and PMM in vitro and in vivo, the occurrence of cross-resistance and the development of resistance upon repeated drug exposure cycles to this drug combination.
Materials and methods
Ethics statement
The use of laboratory rodents was carried out in strict accordance to all mandatory guidelines (EU directives, including the Revised Directive 2010/63/EU on the Protection of Animals used for Scientific Purposes that came into force on 01/01/2013, and the declaration of Helsinki in its latest version) and was approved by the ethical committee of the University of Antwerp, Belgium [UA-ECD 2011–77 (17-02-2012)].
Animals
Female Swiss mice (20–25 g) and female golden hamsters (80–100 g) were purchased from Janvier (France). Food for laboratory rodents (Carfil, Arendonk, Belgium) and drinking water were available ad libitum. Hamsters were kept in quarantine for at least 5 days before infection and were randomly allocated to experimental units of 5 animals each, with the exception of the groups (n = 3) for the selection of resistance.
Leishmania parasites
The L. infantum laboratory strain ITMAP263 (MHOM/MA/67/ITMAP263) was routinely cultivated in Syrian golden hamsters. To infect naive hamsters, ex vivo amastigotes were purified from the spleen of heavily infected donor hamsters, as previously described [14]. Infection inoculates containing 2 x 107 amastigotes/100 μL phosphate buffered saline (PBS) were used to infect hamsters by intracardial injection under isoflurane inhalation anaesthesia. The general condition and body weight of infected animals were monitored daily to evaluate the course of infection. Upon each drug resistance selection cycle, promastigote back-transformation was performed to allow in vitro expansion of surviving parasites for in vitro susceptibility determination purposes.
Promastigotes of the L. infantum clinical isolate LEM3323 (MHOM/FR/96/LEM3323) were obtained from the ‘Centre National de Référence des Leishmania (CNRL)’ (Dr. L. Lachaud) and derived from a French HIV patient. Promastigotes were maintained in HOMEM promastigote medium supplemented with 10% inactivated fetal calf serum (Invitrogen, Ghent, Belgium). To facilitate promastigote back-transformation from infected tissues during in vivo selection of resistance, 20% spent promastigote medium was added and the concentration of inactivated fetal calf serum was augmented to 20% [14].
Drug formulations and preparation
Both MIL (MW = 407.57) and PMM-sulphate (MW = 713.71) were purchased from Sigma (Diegem, Belgium). For the in vitro work, stock solutions of 20 mM were prepared in PBS (MIL) or distilled water (PMM). For the treatment of infected animals, MIL and PMM were formulated in distilled water respectively at 20 mg/mL and 150 mg/mL.
Amastigote susceptibility determination in vitro
In vitro amastigote susceptibilities were determined as previously described [18]. To determine the drug susceptibility of intracellular amastigotes, primary peritoneal macrophages were harvested from starch-stimulated female Swiss mice. Forty-eight hours later, cells were infected with metacyclic promastigotes at an infection ration of 2:1 (LEM3323) or 20:1 (ITMAP236). After 24h, the medium was changed to remove potential extracellular promastigotes. The plates were incubated at 37°C in 5% CO2 atmosphere for another 5 days in presence of 2-fold drug dilutions before staining with Giemsa and microscopic reading.
In vitro efficacy evaluation of MIL-PMM combinations
The normalized fixed ratio isobologram method [19] was used to evaluate the efficacy of the combination of PMM and MIL against the L. infantum clinical isolate (LEM3323) in vitro at their IC50 level. Intracellular amastigotes were exposed to 2-fold dilution series of the drug combinations in fixed drug ratios (5:0, 4:1, 3:2, 2:3, 1:4, 0:5). Top concentrations of MIL and PMM were selected based on their known susceptibility profiles with the intention to center the IC50 of each respective drug in the middle of a seven-point two-fold drug dilution series. For MIL, a top concentration of 5 μM was used while for PMM 500 μM was selected. As in the standard susceptibility assay, drug exposure lasted for 5 days where after the plates were stained with Giemsa and the susceptibility (IC50) towards each drug was determined for each combination ratio. Fractional inhibitory concentrations (FICs) were determined by dividing the IC50 of the drug in combination by the IC50 of the drug alone and were used to construct the isobologram. The sum of FICs (∑FICs) (FIC MIL + FIC PMM) was determined for each fixed-ratio solution and the mean ∑FIC was used to classify the nature of the drug interaction (Fig 1). Interactions are classified synergistic when the mean ∑FIC < 0.5, indifferent when the mean ∑FIC ranges between 0.5 and 4 and antagonistic as the mean ∑FIC > 4.
Fig 1. Isobologram of the interaction of PMM-MIL combination against intracellular amastigotes in vitro at the IC50 level.
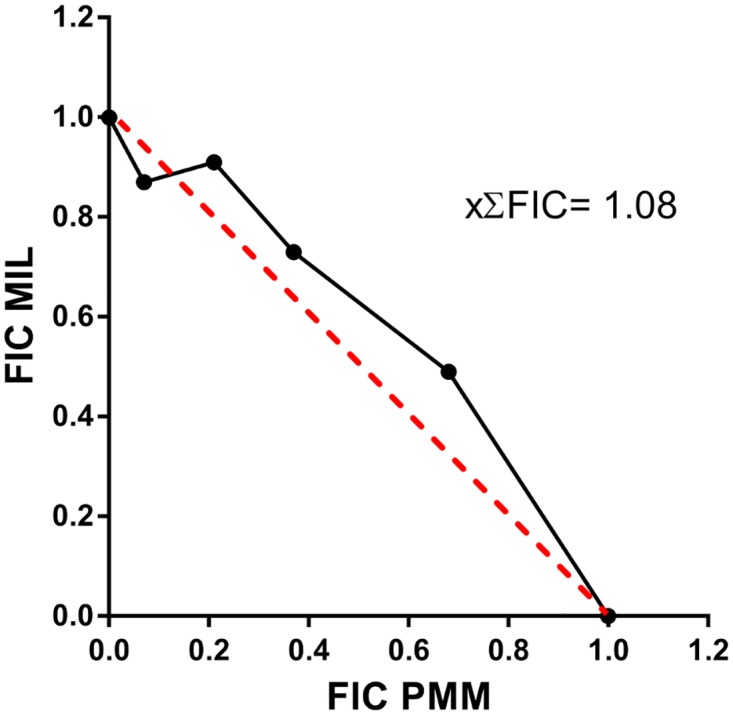
For each fixed-ratio, normalized fractional inhibitory concentrations (FICs) are presented for MIL on the y-axis and for PMM on the x-axis. The constructed isobologram is the result of 4 independent experiments run in duplicate.
In vivo efficacy evaluation of MIL-PMM combination therapy
Before defining the appropriate treatment regimen for the selection of drug resistance in vivo, the efficacy of the envisaged treatment regimens either combined or alone was evaluated. Hamsters were infected with ex vivo ITMAP263 amastigotes as described above. At 21 days post-infection (dpi), animals were treated with either MIL, PMM or the combinations, as listed in Table 1. Due to toxicity, the combination 350 PMM/kg and 40 mg MIL/kg was not included. At 35 dpi, the animals were sacrificed to assess parasite burdens in liver, spleen and bone marrow. Upon fixation in methanol and Giemsa-staining, the liver and spleen imprints and bone marrow smears were evaluated microscopically by counting the number of amastigotes associated with 500 macrophage nuclei. To evaluate drug efficacy, the percentage reduction compared to the untreated control group was calculated.
Table 1. Efficacy of PMM-MIL combination therapy in vivo.
Percentage reduction in amastigote burdens (L. infantum ITMAP263) in liver, spleen and bone marrow compared to the vehicle treated infected control (VIC) group.
| Dosing group: treatment regimen | % reduction of amastigote burdens in target organs | ||
|---|---|---|---|
| Liver | Spleen | Bone-marrow | |
| G1: VIC | - | - | - |
| G2: MIL - 40mg/kg PO—s.i.d. x 5 days | 95.3 | 99.4 | 86.8 |
| G3: MIL—20 mg/kg PO—s.i.d. x 5 days | 80.0 | 94.5 | 76.9 |
| G4: MIL– 10 mg/kg PO—s.i.d. x 5 days | 18.9 | 54.8 | 48.1 |
| G5: PMM—180 mg/kg IP—s.i.d. x 5 days | 79.9 | 64.4 | 0.0 |
| G6: PMM—350 mg/kg IP—s.i.d. x 5 days | 85.1 | 74.4 | 84.5 |
| G7: MIL—20 mg/kg PO—s.i.d. + PMM—350 mg/kg IP—s.i.d. x 5 days | 99.6 | 99.6 | 98.1 |
| G8: MIL—10 mg/kg PO—s.i.d. + PMM—180 mg/kg IP—s.i.d. x 5 days | 97.3 | 96.0 | 88.0 |
| G9: MIL—20 mg/kg PO—s.i.d. + PMM—350 mg/kg IP—s.i.d. x 2 days | 59.3 | 56.1 | 32.5 |
Selection of resistance in vitro
To check whether drug resistance would arise upon PMM-MIL combination, resistance was selected in vitro in the LEM3323 isolate as described earlier [13]. The highest combined drug concentrations were 500 μM PMM and 40 μM MIL. The drug susceptibility was evaluated in vitro after each selection cycle. The selection process was performed for five successive cycles or until resistance towards PMM (IC50 >150μM) or MIL (IC50 >15μM) was reached [20].
Selection of resistance in vivo
Resistance against the MIL-PMM combination therapy was also selected in vivo, as described earlier [14]. Infected hamsters were repeatedly exposed to sub-curative and sub-toxic doses of the drug combination (i.e. oral administration of MIL at 20 mg/kg and intraperitoneal injection of 350 mg PMM/kg for 2 days). Infected animals were treated for 2 days starting from 21 dpi and closely monitored for treatment relapse. When treatment relapse was suspected, a liver biopsy was taken to quantify the infection burden and to enable susceptibility testing of the drug-exposed parasites upon promastigote back-transformation. Maximum two subsequent treatment cycles were performed in the same animal before ex vivo amastigotes were harvested and transferred to a next naïve animal. Subsequent selection rounds were terminated either when drug susceptibility values indicated a drug-resistant phenotype (IC50 MIL >15μM and IC50 PMM >150μM) or after a maximum of 5 successive treatment/relapse cycles.
Results
In vitro efficacy evaluation of MIL-PMM combinations
For the construction of the isobologram (Fig 1) and determination of drug interaction on the LEM3323 isolate by the applied fixed-ratio isobologram method, four independent assays were performed, each in duplicate. the mean ∑FIC was 1.1 ± 0.3 at the IC50 level with ∑FICs ranging between 0.94 and 1.17, indicating an indifferent interaction between MIL and PMM.
In vivo efficacy evaluation of MIL-PMM combination therapy
Results of the in vivo efficacy study are represented in Table 1. Combination of PMM-MIL for 5 days showed a superior efficacy compared to monotherapy with PMM or MIL at either treatment regimen used. MIL reached higher efficacies in clearing the splenic parasite burden, whereas PMM seemed more efficacious against the hepatic parasite burden. Combination of both drugs resulted in splenic and hepatic reductions of >99% and >98% in the bone marrow at the highest dosage scheme. Shortening the duration of the combination therapy from five to two days resulted in a significant efficacy drop with reductions of <60% in the liver and spleen and only 32.5% in the bone marrow.
Selection of resistance in vitro
Results of the in vitro resistance selection are presented in Table 2. Despite a shift in promastigote back-transformation following exposure to the PMM-MIL combination, allowing collection of surviving promastigotes exposed to increasing drug concentrations, no differences were observed between the susceptibility results of the wild-type strain and the strain that was repeatedly exposed PMM and MIL. Also no cross-resistance could be detected in the MIL and PMM resistant lines.
Table 2. Amastigote susceptibility (IC50) of the in vitro resistance selection procedure to PMM-MIL combination therapy.
No difference was observed between IC50 of the wild-type strain (LEM3323 WT) and the strains that underwent three (LEM3323 PMM/MIL3) or five (LEM3323 PMM/MIL5) repeated exposures to the combined high concentrations of PMM and MIL. Exposure to 5 treatment cycles with either PMM (LEM3323 PMM) or MIL alone (LEM3323 MIL) revealed a clear decrease in drug susceptibility (indicated in bold) [21].
| Strain | Drug susceptibility (μM) | |
|---|---|---|
| Paromomycin (PMM) | Miltefosine (MIL) | |
| LEM3323 WT | 98.0 ± 14.3 | 1.0 ± 0.1 |
| LEM3323 PMM/MIL3 | 109.5 ± 26.0 | 0.7 ± 0.2 |
| LEM3323 PMM/MIL5 | 64.3 ± 13.6 | 0.8 ± 0.4 |
| LEM3323 PMM | 212.6 ± 30.9 | 0.5 ± 0.1 |
| LEM3323 MIL | 68.5 ± 8.3 | > 20.0 |
Selection of resistance in vivo
Five subsequent treatment/relapse cycles were conducted and despite recurrent relapses, no resistant phenotype could be observed for either MIL or PMM (Table 3). In contrast, exposure to PMM alone resulted in a significant decrease in susceptibility against PMM without altered MIL resistance profile.
Table 3. Amastigote susceptibility results of the in vivo resistance selection procedure to PMM-MIL combination therapy.
No difference was observed between IC50 of the wild-type (ITMAP263 WT) and the strain that was exposed five times to high concentrations of PMM and MIL (ITMAP263 PMM/MIL). Exposure to PMM alone (ITMAP263 PMM) revealed a significant susceptibility decrease (indicated in bold)[14]. (ND: not done).
| Strain | Drug susceptibility (μM) | |
|---|---|---|
| Paromomycin (PMM) | Miltefosine (MIL) | |
| ITMAP263 WT | 135.8 ± 41.0 | 2.3 ± 0.3 |
| ITMAP263 PMM/MIL | 123.1 ± 23.6 | 3.2 ± 0.1 |
| ITMAP263 MIL | ND | 3.0 ± 0.3 |
| ITMAP263 PMM | 430.9 ± 24.5 | 3.5 ± 0.4 |
Discussion
In response to the recent development and spread of clinical Sb-resistance in L. donovani, other drugs such as MIL, PMM and AmB have been proposed for VL treatment. Nowadays, a single dose of liposomal AmB has been recommended as first-line treatment in the Indian subcontinent [6], however, despite the initiative from Gilead of donating a large batch of liposomal AmB, the need for temperature-controlled settings still severely limits its widespread use. Moreover and comparable to MIL, the first cases of AmB unresponsiveness have recently been reported [22, 23]. In those endemic areas with large-scale Sb-resistance and an increasing number of MIL and AmB treatment failures, a switch from monotherapy towards combination therapies has been advised whereby the short-term combination of PMM and MIL was already shown to be a safe and efficacious alternative [24]. However, concerns regarding drug resistance have already been raised for both PMM and MIL monotherapy [10–14, 17, 22, 25]. Given these concerns a better understanding of the possible implications of large scale implementation of PMM-MIL combination therapy seems essential. Therefore, the present study aimed at evaluating the efficacy of PMM-MIL combination therapy on L. infantum both in vitro and in the in vivo VL hamster model. Additionally, repeated simultaneous exposure to high doses of PMM and MIL was performed in vivo to evaluate the likelihood of PMM- or MIL-resistance development upon combination therapy.
Given the difficult adaptation of clinical isolates maintained in vitro for some time, the in vivo adapted laboratory strain MHOM/MA/67/ITMAP263 was used for the selection of drug resistance in vivo. For the in vitro work, the clinical isolate LEM3323 was used as this was the only strain so far for which resistance towards both MIL and PMM could already been demonstrated upon monotherapy in vitro [11]. The clear difference in intrinsic susceptibility between both strains is in line with inter-strain variability reported for different parasite species and clinical isolates [26–30]. Although the selection of MIL resistance on amastigotes proved to be very challenging in vitro, it could be hypothesized that the enhanced fitness profile associated with PMM resistance could facilitate development of resistance towards other drugs [17]. Our susceptibility analyses did not reveal any direct cross-resistance between PMM and MIL in the individual resistant lines. Despite the fairly rapid selection of PMM resistance upon monotherapy both in vitro and in vivo [11, 14], no decrease in drug susceptibility could be observed when PMM was administered in combination with MIL either in vitro or in vivo. This contrasts with the findings of Garcia-Hernandez et al., who were able to select resistance to MIL-PMM combinations on L. donovani promastigotes [31]. This discrepancy is undoubtedly linked to the stage-dependent outcome of the selection procedure for PMM where selection on amastigotes proved to result in parasites with a susceptible promastigote phenotype [13], indicating that the induced mechanisms of resistance are probably different. Contrary to our previous in vivo resistance selection experiments during which the parasites were exposed to monotherapy of PMM at 350 mg/kg/day x 5 days or to MIL at 20 mg/kg/day x 5 days [14], the dose regimen in the present study (PMM at 350 mg/kg/day and MIL at 20 mg/kg/day x 2 days) proved to be poorly effective with only 59.3% reduction of parasite burden in the liver, 56.1% in the spleen and 32.5% in the bone marrow (Table 1). Comparable to the field situation where treatment duration was reduced from 28 days MIL at 100 mg/kg/day [32] or 21 days PMM at 11 mg/kg/day [33] to 10 days combination therapy (MIL 100 mg/kg/day + PMM 15 mg/kg/day) [8], we also opted to reduce the treatment duration by about half in the current experimental design. While this resulted in better efficacy in man [8], this approach was clearly inferior to drug monotherapy in the Syrian golden hamster model. On the contrary, combining sub-curative doses of both drugs while maintaining the same treatment duration as in monotherapy resulted in an enhanced efficacy, which is in agreement with previous reports [34]. The relatively moderate in vivo activity of PMM at near-toxic doses (350 mg/kg/day) reported here (85.1% reduction in liver, 74.4% in spleen and 84.5% in bone marrow) was already reported previously [14, 35]. Unlike the respective monotherapies, the combination therapy resulted in equally efficient reductions of splenic and hepatic parasite burdens. Although the latter combined dosing schedules showed an excellent efficacy, their high efficacy would severely delay the onset of relapse and complicate repeated relapse/treatment cycles.
The in vitro interaction between PMM and MIL on the recent clinical L. infantum isolate LEM3323 seemed to be indifferent, corroborating observations made for L. donovani [35]. Although both a MIL- and a PMM resistant phenotype could be generated in vitro within 5 drug selection cycles [13, 21], no shift in susceptibility was observed upon in vitro drug combination exposure (Table 2). Similar to MIL resistance selection on other strains, a shift in promastigote back-transformation could be observed, indicating that parasites were able to resist elevated drug concentrations [11]. In case of MIL monotherapy, this observation could not be linked to phenotypic parasite changes in drug susceptibility or changes in treatment outcome [36], leaving its clinical relevance still debatable.
Despite several concerns in the past, the results of this study support the view that MIL-PMM combination therapy can be an effective and appealing choice of treatment in view of the fact that the anticipated rapid development of PMM resistance appears to be delayed in combination treatment with MIL.
Acknowledgments
The authors wish to thank Dr. Lachaud for kindly providing strain LEM3323. Further thanks to Pim-Bart Feijens and Mandy Vermont for their excellent technical assistance with the laboratory and animal work.
Data Availability
All relevant data are within the paper.
Funding Statement
This work was funded by the Fonds Wetenschappelijk Onderzoek (www.fwo.be), grant numbers G051812N (LM) and 12I0317N (SH). The work was also supported by a research fund of the University of Antwerp (www.uantwerpen.be), grant number TT-ZAPBOF 33049 (GC). LMPH is a partner of the Antwerp Drug Discovery Network (ADDN, www.addn.be) and the Excellence Centre ‘Infla-Med’ (www.uantwerpen.be/infla-med). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Ready PD. Epidemiology of visceral leishmaniasis. Clin Epidemiol. 2014;6:147–54. 10.2147/CLEP.S44267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Monge-Maillo B, Lopez-Velez R. Therapeutic options for visceral leishmaniasis. Drugs. 2013;73(17):1863–88. Epub 2013/10/31. 10.1007/s40265-013-0133-0 ; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chakravarty J, Sundar S. Drug resistance in leishmaniasis. J Glob Infect Dis. 2010;2(2):167–76. 10.4103/0974-777X.62887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dhillon GP, Sharma SN, Nair B. Kala-azar elimination programme in India. J Indian Med Assoc. 2008;106(10):664, 6–8. [PubMed] [Google Scholar]
- 5.Jha RK, Sah AK, Shah DK, Sah P. The treatment of visceral leishmaniasis: safety and efficacy. JNMAJ Nepal Med Assoc. 2013;52(192):645–51. [PubMed] [Google Scholar]
- 6.Sundar S, Chakravarty J, Agarwal D, Rai M, Murray HW. Single-dose liposomal amphotericin B for visceral leishmaniasis in India. New Engl J Med. 2010;362(6):504–12. Epub 2010/02/12. 10.1056/NEJMoa0903627 . [DOI] [PubMed] [Google Scholar]
- 7.Mondal D, Alvar J, Hasnain MG, Hossain MS, Ghosh D, Huda MM, et al. Efficacy and safety of single-dose liposomal amphotericin B for visceral leishmaniasis in a rural public hospital in Bangladesh: a feasibility study. Lancet Glob Health. 2014;2(1):e51–7. Epub 2014/08/12. 10.1016/S2214-109X(13)70118-9 . [DOI] [PubMed] [Google Scholar]
- 8.Sundar S, Sinha PK, Rai M, Verma DK, Nawin K, Alam S, et al. Comparison of short-course multidrug treatment with standard therapy for visceral leishmaniasis in India: an open-label, non-inferiority, randomised controlled trial. Lancet. 2011;377(9764):477–86. Epub 2011/01/25. 10.1016/S0140-6736(10)62050-8 . [DOI] [PubMed] [Google Scholar]
- 9.Dorlo TP, Balasegaram M, Beijnen JH, de Vries PJ. Miltefosine: a review of its pharmacology and therapeutic efficacy in the treatment of leishmaniasis. J Antimicrob Chemother. 2012;67(11):2576–97. 10.1093/jac/dks275 [DOI] [PubMed] [Google Scholar]
- 10.Cojean S, Houze S, Haouchine D, Huteau F, Lariven S, Hubert V, et al. Leishmania resistance to miltefosine associated with genetic marker. Emerg Infect Dis. 2012;18(4):704–6. 10.3201/eid1804.110841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hendrickx S, Boulet G, Mondelaers A, Dujardin JC, Rijal S, Lachaud L, et al. Experimental selection of paromomycin and miltefosine resistance in intracellular amastigotes of Leishmania donovani and L. infantum. Parasitol Res. 2014;113(5):1875–81. 10.1007/s00436-014-3835-7 [DOI] [PubMed] [Google Scholar]
- 12.Srivastava S, Mishra J, Gupta AK, Singh A, Shankar P, Singh S. Laboratory confirmed miltefosine resistant cases of visceral leishmaniasis from India. Parasites & vectors. 2017;10(1):49 Epub 2017/02/01. 10.1186/s13071-017-1969-z ; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hendrickx S, Inocencio da Luz RA, Bhandari V, Kuypers K, Shaw CD, Lonchamp J, et al. Experimental induction of paromomycin resistance in antimony-resistant strains of L. donovani: outcome dependent on in vitro selection protocol. PLoS Negl Trop Dis. 2012;6(5):e1664 10.1371/journal.pntd.0001664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hendrickx S, Mondelaers A, Eberhardt E, Delputte P, Cos P, Maes L. In Vivo Selection of Paromomycin and Miltefosine Resistance in Leishmania donovani and L. infantum in a Syrian Hamster Model. Antimicrob Agents Chemother. 2015;59(8):4714–8. 10.1128/AAC.00707-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maarouf M, Adeline MT, Solignac M, Vautrin D, Robert-Gero M. Development and characterization of paromomycin-resistant Leishmania donovani promastigotes. Parasite. 1998;5(2):167–73. 10.1051/parasite/1998052167 [DOI] [PubMed] [Google Scholar]
- 16.Bhandari V, Sundar S, Dujardin JC, Salotra P. Elucidation of cellular mechanisms involved in experimental paromomycin resistance in Leishmania donovani. Antimicrob Agents Chemother. 2014;58(5):2580–5. 10.1128/AAC.01574-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hendrickx S, Beyers J, Mondelaers A, Eberhardt E, Lachaud L, Delputte P, et al. Evidence of a drug-specific impact of experimentally selected paromomycin and miltefosine resistance on parasite fitness in Leishmania infantum. J Antimicrob Chemother. 2016. Epub 2016/04/17. 10.1093/jac/dkw096 . [DOI] [PubMed] [Google Scholar]
- 18.Vermeersch M, da Luz RI, Tote K, Timmermans JP, Cos P, Maes L. In vitro susceptibilities of Leishmania donovani promastigote and amastigote stages to antileishmanial reference drugs: practical relevance of stage-specific differences. Antimicrob Agents Chemother. 2009;53(9):3855–9. 10.1128/AAC.00548-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seifert K, Croft SL. In vitro and in vivo interactions between miltefosine and other antileishmanial drugs. Antimicrob Agents Chemother. 2006;50(1):73–9. Epub 2005/12/27. 10.1128/AAC.50.1.73-79.2006 ; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maes L, Cos P, Croft S. The relevance of susceptibility tests, breakpoints and markers In: Ponte-Sucre A, Diaz E, Padrón-Nieves M, editors. Drug Resistance in Leishmania Parasites: Springer; Vienna; 2013. p. 407–29. [Google Scholar]
- 21.Hendrickx S, Mondelaers A, Eberhardt E, Lachaud L, Delputte P, Cos P, et al. Intracellular amastigote replication may not be required for successful in vitro selection of miltefosine resistance in Leishmania infantum. Parasitol Res. 2015;114(7):2561–5. 10.1007/s00436-015-4460-9 [DOI] [PubMed] [Google Scholar]
- 22.Rijal S, Ostyn B, Uranw S, Rai K, Bhattarai NR, Dorlo TP, et al. Increasing failure of miltefosine in the treatment of kala-azar in Nepal and the potential role of parasite drug resistance, reinfection, or noncompliance. Clin Infect Dis. 2013;56(11):1530–8. 10.1093/cid/cit102 [DOI] [PubMed] [Google Scholar]
- 23.Purkait B, Kumar A, Nandi N, Sardar AH, Das S, Kumar S, et al. Mechanism of amphotericin B resistance in clinical isolates of Leishmania donovani. Antimicrob Agents Chemother. 2012;56(2):1031–41. 10.1128/AAC.00030-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sundar S, Sinha PK, Rai M, Verma DK, Nawin K, Alam S, et al. Comparison of short-course multidrug treatment with standard therapy for visceral leishmaniasis in India: an open-label, non-inferiority, randomised controlled trial. Lancet. 2011;377(9764):477–86. 10.1016/S0140-6736(10)62050-8 [DOI] [PubMed] [Google Scholar]
- 25.Fernandez-Prada C, Vincent IM, Brotherton MC, Roberts M, Roy G, Rivas L, et al. Different Mutations in a P-type ATPase Transporter in Leishmania Parasites are Associated with Cross-resistance to Two Leading Drugs by Distinct Mechanisms. PLoS Negl Trop Dis. 2016;10(12):e0005171 Epub 2016/12/03. 10.1371/journal.pntd.0005171 ; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yardley V, Croft SL, De DS, Dujardin JC, Koirala S, Rijal S, et al. The sensitivity of clinical isolates of Leishmania from Peru and Nepal to miltefosine. Am J Trop Med Hyg. 2005;73(2):272–5 [PubMed] [Google Scholar]
- 27.Morais-Teixeira E, Damasceno QS, Galuppo MK, Romanha AJ, Rabello A. The in vitro leishmanicidal activity of hexadecylphosphocholine (miltefosine) against four medically relevant Leishmania species of Brazil. Mem Inst Oswaldo Cruz. 2011;106(4):475–8 [DOI] [PubMed] [Google Scholar]
- 28.Coelho AC, Trinconi CT, Costa CH, Uliana SR. In vitro and in vivo miltefosine susceptibility of a Leishmania amazonensis isolate from a patient with diffuse cutaneous leishmaniasis. PLoS Negl Trop Dis. 2014;8(7):e2999 Epub 2014/07/18. 10.1371/journal.pntd.0002999 ; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fernandez OL, Diaz-Toro Y, Ovalle C, Valderrama L, Muvdi S, Rodriguez I, et al. Miltefosine and antimonial drug susceptibility of Leishmania Viannia species and populations in regions of high transmission in Colombia. PLoS Negl Trop Dis. 2014;8(5):e2871 Epub 2014/05/24. 10.1371/journal.pntd.0002871 ; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Obonaga R, Fernandez OL, Valderrama L, Rubiano LC, Castro Mdel M, Barrera MC, et al. Treatment failure and miltefosine susceptibility in dermal leishmaniasis caused by Leishmania subgenus Viannia species. Antimicrob Agents Chemotherapy. 2014;58(1):144–52. Epub 2013/10/23. 10.1128/aac.01023-13 ; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garcia-Hernandez R, Manzano JI, Castanys S, Gamarro F. Leishmania donovani develops resistance to drug combinations. PLoSNeglTropDis. 2012;6(12):e1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sundar S, Olliaro PL. Miltefosine in the treatment of leishmaniasis: Clinical evidence for informed clinical risk management. Ther Clin Risk Manag. 2007;3(5):733–40. [PMC free article] [PubMed] [Google Scholar]
- 33.Sundar S, Jha TK, Thakur CP, Sinha PK, Bhattacharya SK. Injectable paromomycin for Visceral leishmaniasis in India. N Engl J Med. 2007;356(25):2571–81. 10.1056/NEJMoa066536 [DOI] [PubMed] [Google Scholar]
- 34.Sane SA, Shakya N, Gupta S. Immunomodulatory effect of picroliv on the efficacy of paromomycin and miltefosine in combination in experimental visceral leishmaniasis. Exp Parasitol. 2011;127(2):376–81. 10.1016/j.exppara.2010.09.003 [DOI] [PubMed] [Google Scholar]
- 35.Seifert K, Croft SL. In vitro and in vivo interactions between miltefosine and other antileishmanial drugs. Antimicrob Agents Chemother. 2006;50(1):73–9. 10.1128/AAC.50.1.73-79.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hendrickx S, Eberhardt E, Mondelaers A, Rijal S, Bhattarai NR, Dujardin JC, et al. Lack of correlation between the promastigote back-transformation assay and miltefosine treatment outcome. J Antimicrob Chemother. 2015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are within the paper.