

Abstract
In this study, we demonstrate that mice deficient in TNFR1 (TNFR1-/-) were resistant to LPS-induced encephalopathy. Systemic administration of lipopolysaccharide (LPS) induces a widespread inflammatory response similar to that observed in sepsis. Following LPS administration TNFR1-/- mice had less caspase-dependent apoptosis in brain cells and fewer neutrophils infiltrating the brain (p<0.039), compared to control C57Bl6 (TNFR1+/+) mice. TNFR1-dependent increase in aquaporin (AQP)-4 mRNA and protein expression was observed with a concomitant increase in water content, in brain (18% increase in C57Bl6 mice treated with LPS vs those treated with saline), similar to cerebral edema observed in sepsis. Furthermore, absence of TNFR1 partially but significantly reduced the activation of astrocytes, as shown by immunofluorescence and markedly inhibited iNOS mRNA expression (p<0.01).
Septic Encephalopathy is a devastating complication of sepsis. Although, considerable work has been done to identify the mechanism causing the pathological alterations in this setting, the culprit still remains an enigma. Our results demonstrate for the first time that endotoxemia leads to inflammation in brain, with alteration in blood-brain barrier, up-regulation of AQP4 and associated edema, neutrophil infiltration, astrocytosis, as well as apoptotic cellular death, all of which appear to be mediated by TNF-α signaling through TNFR1.
Keywords: Rodent, Septic Encephalopathy, Lipopolysaccharide, Apoptosis, Aquaporin 4 Neuroimmunology
Sepsis is a condition characterized by uncontrolled infection and affects many organs including brain (Green et al., 2004), with an attendant high mortality rate. Administration of bacterial endotoxin (LPS), a cell wall component of gram-negative bacteria, to mice cause pathogenesis, mimicking what occurs in clinical sepsis (Gardenfors et al., 2002;Radzivil et al., 1990). Septic encephalopathy could be due to multiple factors including inflammatory cells and their mediators, reduced cerebral blood flow, disruption of the blood-brain barrier (BBB), cerebral edema and inflammation (Papadopoulos et al., 2000). One of the mediators of inflammation that play a key role in sepsis, tumor necrosis factor (TNF-α), is increased in circulation following LPS administration (Tsao et al., 2001;Merrill and Benveniste, 1996). Serum TNF-α is an acute phase response and does not correlate with the alterations in brain, during stroke (Intiso et al., 2004). On the other hand, TNF-α is also produced in brain, by microglia and astrocytes, both of which are activated by LPS. LPS disrupts the BBB (Tsao et al., 2001;Gurney et al., 2006), which leads to increased infiltration of blood cells into brain. TNF-α has also been shown to stimulate the oxidative burst of neutrophils (Granert et al., 1994;Wispelwey et al., 1989) and to up-regulate the expression of adhesion molecules (Amrani et al., 2000;Marshall et al., 2003;Schafers et al., 2002;Yang et al., 2005) that mediate neutrophil recruitment to tissues following an inflammatory stimulus. TNF-α participates in many pathophysiological conditions in the central nervous system (CNS) (Pan et al., 1997b), including neurodegenerative diseases (Alexander et al., 2005;Alexander et al., 2007). The significance of TNF-α in these neuropathological conditions is not totally understood, since it has potentially both detrimental and beneficial roles in the CNS. The role of TNF-α is manifold and range from apoptosis and astrogliosis, a prominent feature of inflammation in brain, to the regulation of iNOS synthesis and edema. On the other hand, several lines of evidence show that TNF-α can also exhibit regenerating or neuroprotective effects (Loddick and Rothwell, 1999;Allan and Rothwell, 2001). TNF-α acts through two distinct receptors: TNFR1, which mediates most of the inflammatory actions of TNF-α, and TNFR2, which has been proposed to have an anti-inflammatory role (Dopp et al., 1997). Although both TNF-α receptors occur in brain, TNFR1 seems to be abundantly expressed in the brain and shows constitutive expression in astrocytes (Dopp et al., 1997) and the microvasculature (Nadeau and Rivest, 1999), while TNFR2 is observed only on stimulation in brain.
Astrocytes are the major glial cell population within the CNS and play important physiological roles in brain functions. Astrocytes react to various neurodegenerative insults and diseases, leading to vigorous astrogliosis (Eng et al., 1992). On activation, astrocytes express an enhanced level of glial fibrillary acidic protein (GFAP) proportional to the severity of astroglial activation (Eng et al., 2000;Eng and Ghirnikar, 1994;Eng et al., 1992). However, the mechanism by which astroglial expression of GFAP is increased in neurodegenerative CNS remains unclear (Norenberg, 1996;Norenberg, 1998). Several lines of evidence demonstrate that NO plays a key role in regulating the expression of GFAP in astrocytes (Askalan et al., 2006;Enkhbaatar et al., 2006;Hawkins et al., 1998). On the other hand activated astrocytes express inducible nitric oxide (NO) synthase (iNOS) leading to an excessive amount of NO, a molecule implicated in many neurodegenerative and neuroinflammatory conditions (Lee et al., 2004;Choi et al., 2005;Kubes et al., 1993). NO could induce apoptosis in astrocytes by p-53 and Bax-dependent mechanisms (Yung et al., 2004). Furthermore, aquaporin (AQP) 4, the most abundant water channel in the CNS is present on the astrocytes (Vizuete et al., 1999;Rao et al., 2003) and plays a crucial role in brain edema formation observed after CNS insults or in disease (Manley et al., 2004;Badaut et al., 2002;Alexander et al., 2003). Edema could lead to increased intracranial pressure, resulting in brain herniation and finally death (Norenberg et al., 2005).
Septic encephalopathy although common and devastating, the role of the different mechanisms behind this occurrence, still remains unclear. Although cytokines such as TNF-α are key mediators of sepsis (Lucas et al., 1997), knowledge as to their effects in brain is still being investigated, in the setting of endotoxemia. As many of the central consequences of TNF-α are mediated by the TNFR1 receptor (Gimenez et al., 2004b;Yin et al., 2004a), the present investigation attempts to gain further insights into the role of this receptor in endotoxin induced toxicity, in brain.
Experimental Procedures
Animals
Mice with targeted deletion of TNFR1 (TNFR1-/-) were obtained from The Jackson Laboratory (Bar Harbor, ME) and used for experiments at 8 wk of age. Because these mice are on a C57BL/6 background, age-matched C57BL/6 mice were used as controls. The animals were maintained with 12 h light and dark cycles with free access to food and water. One group was given LPS (from Escherichia coli, serotype 055:B55, Sigma Aldrich, St. Louis, MO., Lot # 127H4097) as a single injection of 0.15 mg i.p. and another group received the saline vehicle as control. The mice were sacrificed 8 h later and their brains harvested. The animals were maintained and experiments were performed in accordance with the guidelines set by the University of Chicago Institutional Animal Care and Use Committee.
Tissue processing
Brains were isolated from animals immediately upon sacrifice. Cerebellum and brain stem were discarded. The anterior region of the cerebral cortex was snap frozen for immunofluorescence (IF) microscopy, and the rest were processed for RNA and genomic DNA isolation. For immunohistochemistry, 4-μm brain cryostat sections were fixed with ether/ethanol, incubated with 0.3% H2O2 for 30 min, and blocked with dilute horse serum. Sections were stained for neutrophils by sequential incubation with rat anti-mouse neutrophil (mAb 7/4; Serotec, Raleigh, NC) at a 1/60 dilution for 30 min followed by HRP-conjugated rabbit anti-rat IgG (Sigma-Aldrich) at a 1/60 dilution for 30 min and diaminobenzidine reagent (Vector Laboratories, Burlingame, CA) for 10 min. A blinded observer counted the number of neutrophils per high-power field and recorded the average of 10 fields for each sample.
LM-PCR
Brains were harvested and immediately frozen at -80°C. Later, a small piece was thawed, from which DNA was purified (DNeasy DNA purification system; Qiagen, Valencia, CA). The extent of DNA laddering was amplified and detected by ligase-mediated (LM)-PCR (Clontech Laboratories, Palo Alto, CA) according to the manufacturer's instructions, as follows. DNA isolated from each animal was incubated with the supplied primer targets and T4 DNA ligase for 18 h at 16°C. Twenty micrograms of this ligated DNA was then used as the substrate for PCR, using supplied primers and Advantage DNA polymerase (Clontech Laboratories) for 23 cycles of 94°C for 1 min and at 72°C for 3 min. The reaction product for each animal was run on a 1.3% agarose gel, and ethidium bromide-stained bands were detected with UV light illumination.
Assay of caspase-3 activity
Brains were homogenized in cell lysis buffer (25 mM HEPES pH 7.4, 2 mM DTT, 5 mM EDTA, and 10 mM digitonin). Lysates were then incubated on ice for 15 min and centrifuged at 10,000 g for 10 min at 4°C and protein concentrations were determined using a BCA assay. Lysates (soluble supernatants) were used immediately or stored at -80°C. Aliquots of protein (50μg) were incubated at 37°C with assay buffer (50 mm HEPES pH 7.4, 100 mm NaCl, 0.1% CHAPS, 10 mm dithiothreitol, 1 mM EDTA, 10% glycerol) and 200 mM Ac-DEVD-pNA (Biomol). Hydrolysis of the DEVD-AFC substrate was followed for 15 min by fluorimetry of the released AFC (excitation 400 nm, emission 505 nm) and activity calculated from the slope.
Brain water content
We determined the brain water content in normal and LPS-treated mice. The brains were removed, weighed immediately and then kept at 300°C for 48 h. The brains were weighed during this process at 24 h and then again at 48 h to make sure they had reached a consistent weight. The percentage of brain water in the tissue was calculated as (wet wt – dry wt) * 100/wet wt.
Blood-Brain barrier integrity
The BBB integrity was determined in normal, LPS-treated C57Bl6 and TNFR1-deficient mice. 1% Evans blue in saline was injected through the tail vein and the mice were sacrificed 5 min later. The brains were removed, snap frozen. Cryosections (4μm thickness) were obtained and observed using a Zeiss microscope at 40×.
IF microscopy
Cryostat sections (4μm) were fixed with ethanol:ether and ethanol and IF microscopy was done using hamster anti-mouse TNFR1 (BD PharMingen, San Diego, CA), rabbit anti-rat AQP4 (Chemicon International, Temecula, CA) and rabbit anti-cow GFAP (Dako) at dilutions of 1:60. The staining was detected using FITC-labeled anti-hamster or anti-rabbit antibody at a dilution of 1:100. No staining was observed in controls slides treated with a rat or rabbit IgG followed by FITC-labeled anti-rabbit antibody. Sections were observed using a Zeiss microscope at 400×. The slides were scored in a blinded fashion from 0-4 (0 indicating no staining and 4 the most intense staining).
Quantitative (q) RT-PCR
RNA from brains was isolated using TriZol reagent (Life Technologies, Grand Island, NY). cDNA was prepared in an analogous fashion from brains of mice (TNFR1-/- and TNFR1+/+) sacrificed 8 h after LPS injection and compared with C57Bl6 mice treated with saline (n = 6 per group). Real-time RT-PCR was performed for iNOS, and AQP4 as follows. A portion of brain from each mouse was frozen at -80°C, from which total RNA was purified using the RNeasy Mini RNA purification kit (Qiagen). To remove all traces of genomic DNA, samples were then treated with RNase-free RQ1 DNase (1 U per 4 μg RNA; Promega, Madison, WI) in 10 μl reaction buffer (final concentration, 40 mM Tris-HCl, 10 mM MgSO4, and 1 mM CaCl2 (pH 8)) at 37°C for 30 min. This was followed by addition of 1 μl 20 mM EGTA (pH 8) to stop the reaction and incubation at 65°C for 10 min to inactivate the DNase. cDNA was generated from RNA using random hexamers as primers with the SuperScript first-strand synthesis kit (Life Technologies), according to the manufacturer's instructions. Real-time PCR was performed using a Smart Cycler (Cepheid, Sunnyvale, CA). For iNOS and GAPDH measurements, probes were labeled at the 5′ end with the reporter dye molecule FAM (6-carboxyfluorescein; emission λmax = 518 nm) and at the 3′ end with the quencher dye molecule TAMRA (6-carboxytetramethyl-rhodamine; emission λmax = 582 nm). Each reaction was conducted in a total volume of 25 μl with l× TaqMan Master Mix (PE Applied Biosystems, Foster City, CA), 3 μl sample or standard cDNA, primers at 200 nM each, and probe at 100 nM. PCR was conducted with a hot start at 95°C (5 min), followed by 45 cycles of 95°C for 15 s and 60°C for 30 s. For each sample, the number of cycles required to generate a given threshold signal (Ct) was recorded. Using a standard curve generated from serial dilutions of splenic cDNA, the ratio of iNOS expression relative to GAPDH expression was calculated for each experimental animal and normalized relative to an average of ratios from TNFR1+/+ brain unexposed to LPS. Measurements of TNF-α TNFR1 and AQP4 mRNA expression relative to GAPDH were performed in an analogous fashion, but using the SybrGreen intercalating dye method with HotStar DNA polymerase (PE Applied Biosystems) instead of labeled probes. Reaction conditions were according to the manufacturer's instructions. The reaction temperature profiles for TNF-α, TNFR1 and AQP4 were as described above, except that the annealing temperature was 58°C.
Statistics
Data were analyzed with Minitab software (State College, PA). Unless noted otherwise, data are given as the mean ± SEM. Groups were compared by two-sample t test. A value of p ≤0.05 was considered significant.
Results
TNF-α mediates septic encephalopathy by acting on its receptor, TNFR1 in brain
TNF-α is released systemically into circulation after LPS administration (Fig 1). TNF-α could mediate LPS-induced septic encephalopathy, either indirectly by stimulating the release of other proinflammatory cytokines or by acting directly on its receptor, TNFR1 in brain (Schafers et al., 2002; Merrill and Benveniste, 1996). Therefore, to determine the role of TNF-α and its receptor, TNFR1 as mediators in septic encephalopathy, it was important to know whether their expression was altered in endotoxemia. Using real-time RT-PCR, we found both TNF-α and TNFR1 mRNA to be increased in brain by a factor of 6 and 3 after LPS administration compared to saline-treated controls (n = 6 per group; p < 0.001) (Fig 2). Furthermore, by IF, TNFR1 expression was significantly elevated in endotoxin treated mice, especially in the hippocampal region (Fig 3 A). TNFR1-/- brain sections served as negative controls. As reported earlier, TNFR1 is expressed on the surface of the astrocytes (Fig 3 B). Shown is immunofluorescence (IF) micrographs of paraformaldehyde-fixed cortical sections stained with (A) GFAP or (B) TNFR1 and (C) merge.
FIGURE 1. TNF-α is significantly increased in circulation by LPS-treatment.
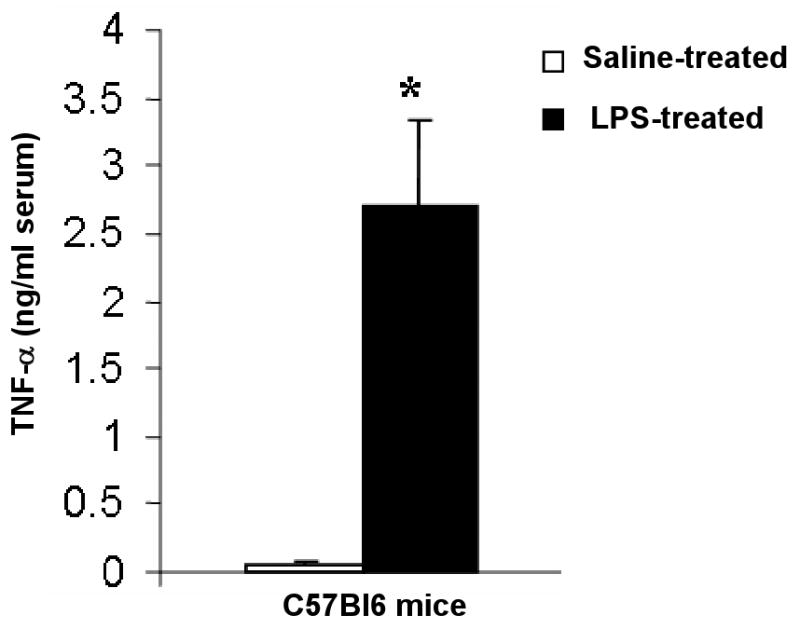
Serum TNF-α was determined by ELISA. At baseline, circulating levels of TNF-α were undetectable using a sensitive ELISA technique. Two h after LPS injection, there was a profound (25 fold) increase in serum TNF-α. n = 6 per group. *p < 0.001 vs saline-treated.
FIGURE 2. Upregulated brain mRNA expression of TNF-α and its receptor, TNFR1 in endotoxemia.

mRNA expression of TNF-α and TNFR1 were quantified by qRT-PCR and normalized to expression of GAPDH. Systemic administration of LPS caused a significant increase in expression of TNF- α (7 fold) and TNFR1 (4 fold), n = 6 per group. *p < 0.01 vs. saline-treated.
FIGURE 3.
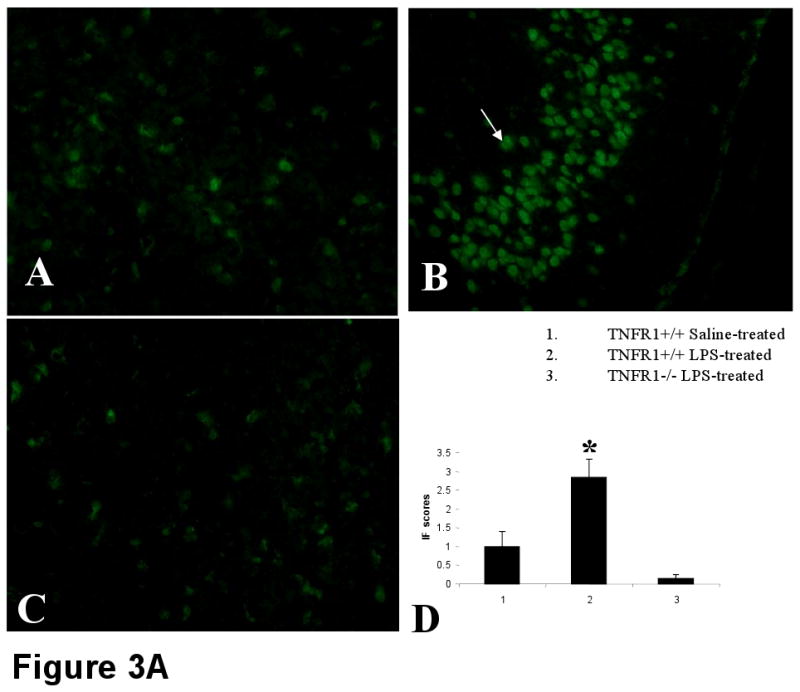
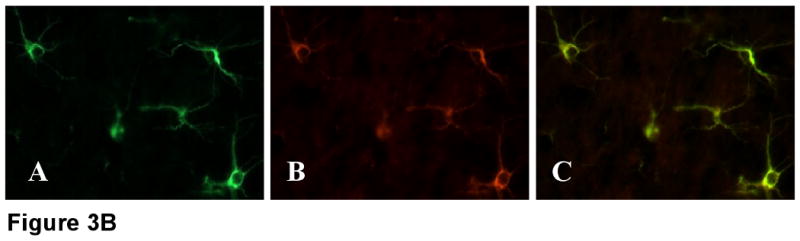
FIGURE 3A. LPS administration significantly up regulates TNFR1 expression, by immunofluorescence.
Representative sections immunostained with anti-TNFR1 followed by FITC-labeled anti-hamster antibody show increased expression of TNFR1 mainly in the hippocampus and cortex of LPS-treated mice (B). Reduced TNFR1 expression was observed in mice receiving saline (A). No TNFR1 expression is apparent in LPS-induced TNFR1-/- mice (C). n=6 in each group. Magnification = 20×. Setting of the microscope was kept strictly unaltered while examining the different groups. The stained sections were scored from 0-4 and the results are given in (D).
FIGURE 3B. Dual staining for GFAP (A) and TNFR1 (B) and merge (C) indicate colocalization. TNFR1 is present on the surface of the astrocytes. Magnification, 40× under oil.
TNFR1 mediates LPS-induced neutrophil infiltration in the brain
Neutrophils have been implicated in the pathogenesis of LPS-induced injury of the brain. TNFR1 was shown to be a major factor involved in the recruitment of inflammatory cells. We confirmed that neutrophils infiltrated the brain following LPS administration, but to a significantly lesser extent in TNFR1-/- mice compared with TNFR1+/+ controls (Fig 4) (2.15 ± 1.3 vs 6.6 ± 1.9 neutrophils per high-power field in TNFR1-/- and TNFR1+/+ mice, respectively; p < 0.01). C57Bl6 mice treated with saline had very few neutrophils in brain (0.65 ± 0.6). This supports a role for TNF-mediated neutrophil recruitment in the pathogenesis of LPS-induced septic shock.
FIGURE 4. Neutrophil accumulation in the LPS-induced brain is TNFR1-dependent.
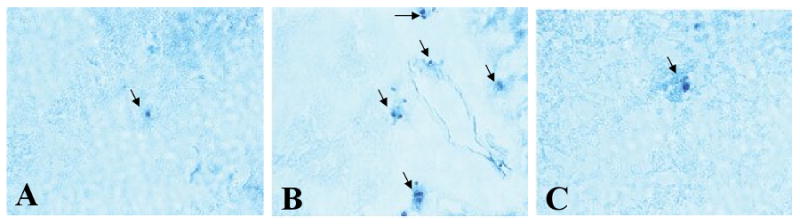
(A) Few neutrophils are present in C57Bl6 brain at baseline (0.65±0.6). (B) Following injection of LPS, marked neutrophil accumulation was found in the brains of C57Bl6 mice, primarily in the cerebral cortex, which is shown (6.6±1.9). (C) TNFR1-/- mice had significantly less number of neutrophils in the brain after LPS injection (2.5±1.3). Magnification, ×200.
LPS causes up-regulation of TNFR1 dependent iNOS expression
iNOS has been shown to be strongly up-regulated in a wide variety of tissues, including the brain, after administration of LPS (Brown and Bal-Price, 2003;Lee et al., 1995;Lee et al., 2004). Previous studies have reported that iNOS activation requires autocrine stimulation by TNF-α (Bi et al., 2005;Marcus et al., 2003). By real-time qRT-PCR, expression of iNOS mRNA in the brain was markedly up-regulated in TNFR1+/+ mice given LPS compared with the saline-treated C57Bl6 mice, (1.2 ± 0.19- and 3.2 ± 0.74-fold increases in control and TNFR1+/+ mice, respectively; p <0.01) and remained closer to normal in the TNFR1-/- mice (Fig 5). Our results indicate that TNF-α induced increased expression of iNOS through its receptor, TNFR1.
FIGURE 5. Expression of iNOS in LPS-induced brain is regulated by TNFR1.

mRNA expression of the inflammatory mediator, iNOS, was quantified by qRT-PCR and normalized to expression of GAPDH (n=6 per group). Significant increase in expression of iNOS (*p < 0.01) was observed in LPS-treated C57Bl6 mice compared to their saline-treated counterparts. Absence of TNFR1 prevented the increase of iNOS expression in LPS-treated brain. Each data point represents an individual animal.
Absence of TNFR1 partially abrogates LPS-induced astrocytosis
Activation of astroglia, also known as astrocytosis, occurs in many neurodegenerative diseases and in response to CNS insults. To determine the role of TNF-α in this CNS response, we assessed GFAP expression in mice sufficient (Fig 6B) and deficient in TNFR1 (Fig 6C) treated with endotoxin and compared them to control C57Bl6 treated with saline (Fig 6A). GFAP expression was significantly increased in mice treated with endotoxin compared to the saline-treated controls. The absence of TNFR1 reduced the increase in GFAP expression but did not prevent it completely indicating that TNF-α may be one of the factors that induce astrogliosis through its receptor, TNFR1.
FIGURE 6. LPS causes activation of astrocytes partially through the actions of TNFR1.
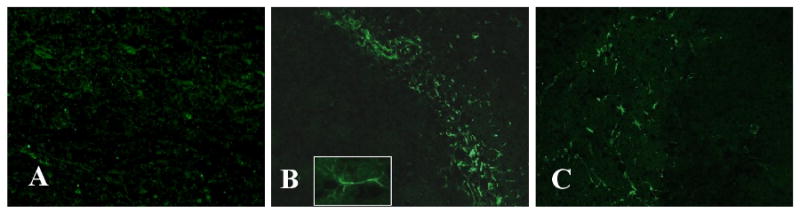
Representative brain sections immunostained with anti-GFAP show activated astrocytes in the hippocampus and cortex of LPS-treated mice (B) while there were virtually no positive cells in those receiving saline (A). Inset is an astrocyte at higher magnification. A marked reduction of these activated cells is apparent in LPS-induced TNFR1-/- mice (C). n=6 in each group. Magnification, 20×.
Mice deficient in TNFR1 are protected against apoptosis that occurs in endotoxin-induced encephalopathy
NO and other cytokines induce apoptosis in neurons which are remarkably sensitive. In endotoxemic mice, there were an increased number of cells in the hippocampus that were shrunken and had condensed cellular structure consistent with ongoing apoptosis (Fig 7). To examine whether TNFR1 regulates apoptosis in brain and to quantify the extent of apoptosis in the brain after systemic LPS administration, LM-PCR was performed to amplify fragmented DNA from brains of C57Bl6 mice treated with saline and C57Bl6 and TNFR1-/- mice treated with endotoxin (Fig 8). Apoptosis was significantly increased in brains from mice treated with endotoxin compared to the saline-treated controls. TNFR1-/- mice were resistant to LPS-induced apoptosis (n = 6 per group). Concomitant with these results, in response to systemic LPS administration caspase-3, the “destroyer enzyme” common to both extrinsic and intrinsic apoptotic pathways was increased 4 fold in brain compared to the saline-treated controls (p<0.01). This increase was significantly attenuated in the absence of TNFR1 (Fig. 9). Therefore, TNF-α, acting through TNFR1, is a key mediator of LPS-induced encephalopathy.
FIGURE 7. Endotoxemia leads to histological changes in the hippocampus.
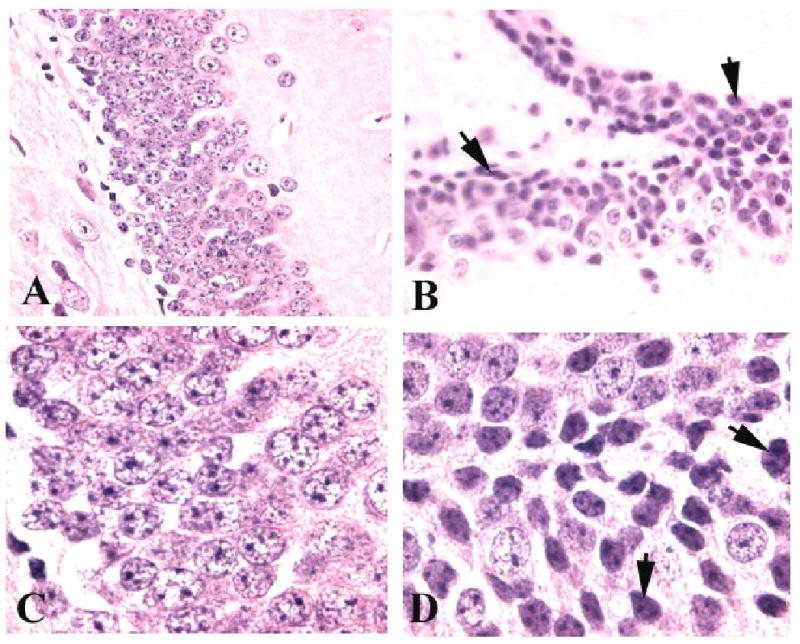
Wild type mice were given saline (A, C) or LPS (B, D) and sacrificed 8 h later for histological analyses. Shown are representative PAS stained sections, from the dentate gyrus of individual mice. Control mice showed normal architecture, while LPS-treated mice had evidence for cellular damage, as indicated by arrows. Magnifications, ×200 (A, C) and 400 (B, D).
FIGURE 8. TNFR1 deficiency significantly reduces endotoxemia-induced apoptosis in brain.
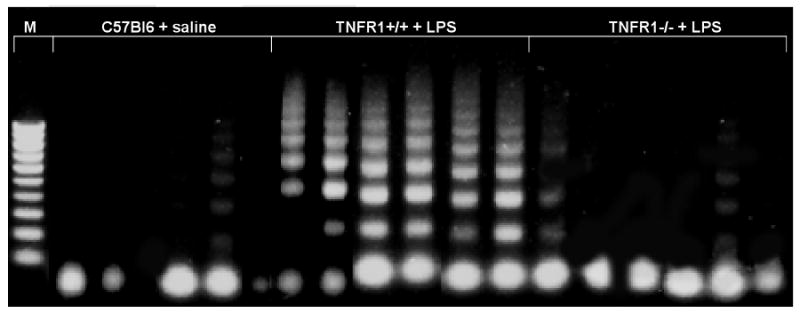
DNA was isolated from brains of C57BL/6 mice treated with saline or 0.15 mg LPS and subjected to LM-PCR and apoptotic DNA laddering was studied. Significant apoptosis was observed in brains of wildtype mice 8 h after LPS injection compared to the negligible amount of apoptosis occurring at baseline (C57Bl6 brain not exposed to LPS). In contrast, the amount of apoptosis after LPS was markedly reduced in the absence of TNFR1. Each lane contains brain tissue harvested from an individual animal.
FIGURE 9. TNFR1 regulates LPS-induced caspase-3 activation in brain.

Caspase- 3 activity was assayed in brains of mice given saline or LPS and sacrificed 8 h later, by means of a fluorogenic assay. LPS administration increased caspase-3 enzyme activity significantly compared with saline-treated control mice. Results are expressed in terms of mM AMC substrate liberated per minute per mg protein. The absence of TNFR1 significantly attenuated this increase, with caspase-3 activity remaining comparable to that of saline-treated controls. Data shown are from individual mice. *p< 0.001 vs. other groups.
Endotoxemia increases brain AQP4 expression and water content
Cerebral edema can occur as a result of a number of factors that include the increased expression of TNF-α. To determine whether edema occurred in brains of mice administered LPS, we measured water content in brains of control and LPS-treated mice. Water content in brain was increased, by approximately 18% in wild-type mice treated with LPS compared to those controls treated with saline (Fig. 10). Edema could be due to both vasogenic and cytotoxic changes. To gain insight into this possibility, we assessed the BBB integrity. Evans blue was observed to leak out of the microcapillaries in the LPS-treated mice forming a halo, indicating that the BBB was compromised (Fig. 11 B) compared to controls (Fig. 11 A) and TNFR1-deficient mice (Fig. 11 C). Since AQP4 is the main water channel protein in brain, we studied its expression in endotoxemic mice. AQP4 expression as determined at the mRNA level by qRT-PCR and at the protein level by IF were significantly increased in LPS-treated mice compared to control mice treated with saline (Fig. 12). The absence of TNFR1 prevented the LPS induced increase in AQP4 expression in brain. Of importance is that the mRNA and protein expression of AQP4 were concordant in all experimental groups.
FIGURE 10. Brain water content is increased in endotoxemia.

Brain water content was measured as described in Methods. Water content of normal brain was taken as 100%. Water content in brains of mice treated with LPS was 18% above the controls. This increase in water was prevented in the absence of TNFR1, 97% of normal mice. *p < 0.01 vs. saline-treated.
FIGURE 11.
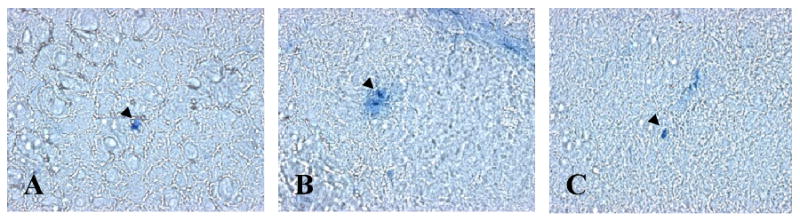
Representative sections of the cerebral cortex from mice perfused with Evans blue are shown. Compared to the controls (A), diffusion of Evans blue from the microcapillaries was observed in the LPS-treated mice (B), indicating possible disruption of the BBB. This diffusion of Evans blue to the surrounding periphery was reduced in TNFR1-/- mice.
FIGURE 12. Increased brain AQP4 expression in endotoxemia is prevented by TNFR1 deficiency.
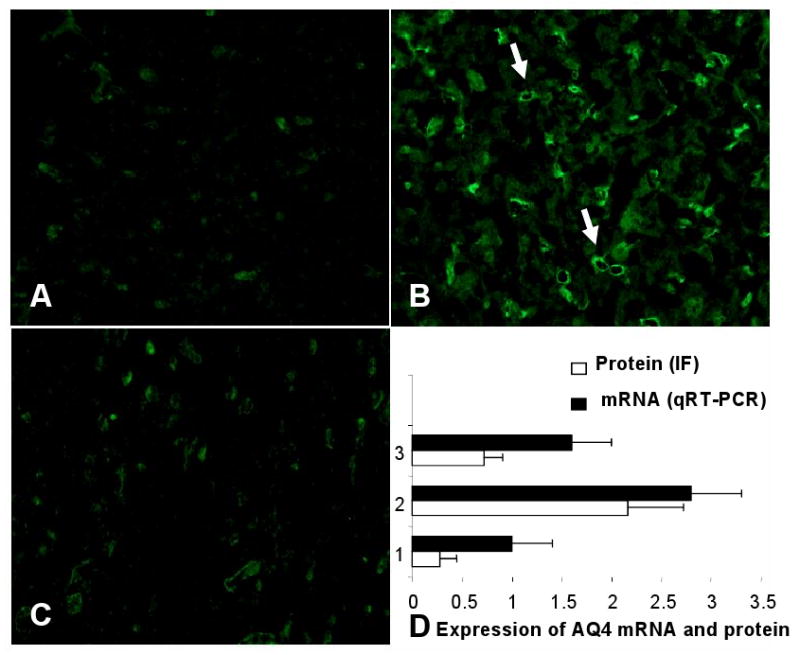
Representative sections of the cerebral cortex stained for AQP4 are shown. Compared to controls given saline (A), there was significantly increased AQP4 expression in wildtype mice given LPS (B). The increase in AQP4 protein was observed especially around the small vessels as indicated by the arrows. This increase was prevented and AQP4 expression remained comparable to saline-treated controls in TNFR1-/- (C) mice. The stained sections were scored from 0-4 and the results are given in D along with mRNA expression quantified by qRT-PCR (n=6 per group). *p < 0.01 vs. other groups.
Discussion
Encephalopathy is a common feature in sepsis occurring in ∼25% of patients often before failure of other organ systems. Patients with septic encephalopathy have a higher mortality rate compared to those without brain involvement, likely reflecting the severity of disease and the direct adverse effects of brain involvement. Given the complexity of sepsis, pathogenesis of septic encephalopathy is multifactorial and includes circulatory and metabolic derangements (Papadopoulos et al., 2000), inflammation (Green et al., 2004), and infections of the brain. Although it has been established that TNF-α is a key mediator in sepsis and the role of TNF-α in LPS-induced encephalopathy has been examined, the exact mechanism still remains an enigma.
TNF-α can induce apoptosis directly through its receptor, TNFR1 that is abundantly present in brain or indirectly through stimulating glial cells to produce neurotoxins such as excitatory amino acids. There are different possible mechanisms by which TNF-α acts through TNFR1 leading to the pathological alterations observed during sepsis, in brain. Activation of TNFR1 could increase permeability of the BBB (Pan et al., 1997a), induce expression of cell adhesion molecules in endothelial cells and astrocytes (Yin et al., 2004b;Gimenez et al., 2004a;Henninger et al., 1997a), thereby favoring leukocyte infiltration and inflammatory brain damage, and can play an important role in inducing apoptosis. After systemic administration of LPS, a profound systemic up regulation of TNF-α was observed, similar to earlier studies (Cunningham et al., 2002). Systemic TNF-α could affect areas with no BBB such as the CVO and in areas where the BBB is breached as a result of LPS-treatment. In addition, TNF-α produced in situ could cause pathological changes. In accord with the results observed in rat brain (Nadeau and Rivest, 1999), mRNA expression of both TNF-α and TNFR1 in our studies were unregulated by i.p. LPS. TNF-α signaling through TNFR1 causes apoptosis via the extrinsic (death receptor) pathway, leading to caspase-8 activation and subsequently the activation of caspase-3 (Hengartner, 2000;Yuan and Yankner, 2000). Our results demonstrate that apoptosis in brains of LPS-treated mice was significantly increased compared to saline-treated controls while little to no apoptosis over baseline occurred in TNFR1-/- mice given LPS. These results indicate, that in this setting of endotoxemia, apoptosis in the brain occurred through the extrinsic pathway in a TNFR1-dependent fashion, possibly in glial cells (Jellinger and Stadelmann, 2000b;Jellinger and Stadelmann, 2000a). However, other known triggers of apoptosis, such as NO, or reactive oxygen intermediates, the latter perhaps derived from infiltrating neutrophils, could be relevant as well. This is an important aspect, since we observed definite disruption of the BBB especially around the microcapillaries, which could alter the movement of circulating cells and water into the brain. Experiments to understand the precise apoptotic pathways involved in LPS-induced encephalopathy, is the subject of ongoing work.
TNF-α, on induction increases the expression of adhesion molecules, ICAM-1 and VCAM and alters the BBB integrity (Henninger et al., 1997b). Since increased expression of ICAM-1 on endothelial cells mediates the infiltration of blood-borne cells into the brain, this could explain the observed increased infiltration of neutrophils into the brain. Infiltration of neutrophils (polymorphonuclear leukocytes) into brain parenchyma could induce activation of astrocytes and promote cerebral inflammation. Activated astrocytes could produce and release TNF-α, and nitric oxide (NO). Furthermore, TNF-α was shown to induce production of NO through TNFR1, in cultured glial cells (Romero et al., 1996;Dong and Benveniste, 2001). iNOS, the protein that catalyzes the formation of NO in injured brain regulates neurodegeneration and cellular apoptosis (Gahm et al., 2006). iNOS mRNA expression was observed to correlate with clinical severity in experimental animals such as models of herpes simplex virus type 1 and experimental allergic encephalitis, suggesting that locally produced NO may play an important pathophysiological role during infection and inflammation of the central nervous system. Consequently, treatments that control the activation of astroglia may be effective at diminishing the severity of LPS-induced encephalopathy.
Astrocytes are an integral part of the blood–brain barrier (BBB), which maintains the internal milieu constant (Davies, 2002;Davson H et al., 1993;Davson H and Segal MB, 1995). Increased expression of the important water channel protein, AQP4 on astrocytes and endothelial cells could lead to the increased water content (Alexander et al., 2003;Badaut et al., 2002;Manley et al., 2004) observed in LPS-induced brains, in our study, similar to edema seen in clinical patients(Papadopoulos et al., 2000). Furthermore, in our study, an 18% increase in brain water content was found in mice that were treated with LPS compared to mice receiving saline, which was reduced in LPS-treated TNFR1-/- mice. Although the increase is significant but not substantial, the brain is located in a defined space and an alteration in the internal milieu could lead to traumatic consequences. In clinical patients with septic encephalopathy the integrity of the BBB is altered with associated edema. Edema is a significant source of morbidity and mortality in several brain disorders and therefore an understanding of its pathophysiology is an important step towards designing therapeutic strategies not only in sepsis but in other CNS diseases too.
In summary, our results demonstrate that TNFR1-mediated signaling is a key mechanism involved in restricting or resolving, the LPS-induced inflammatory response. Absence of the TNF-α/TNFR1 pathway could alleviate apoptosis, reduce neutrophil infiltration, astrogliosis and edema in LPS-induced brain. Additional experiments need to be designed and conducted to understand the extent to which these mechanisms individually contribute to LPS-induced encephalopathy and the extent to which these processes are interrelated. Furthermore, our future studies will address the issue of which populations of cells are dying of apoptosis in this setting. This will lead to a better understanding of the pathophysiology of septic encephalopathy and thereby the designing of more effective therapeutic strategies.
Table 1.
| Gene |
Forward primer 5′-------3′ |
Reverse primer 5′--------3′ |
Probe 5′-------3′ |
|---|---|---|---|
| FAM/TAMRA dyes: | |||
| GAPDH | GGCAAATTCAAGGCACAGT | AGATGGTGATGGGCTTCCC GGCC | GAGAATGGGAAGCTTGTCATC |
| iNOS | CAGCTGGGCTGTACAAACCTT | TGAATGTGATGTTTGCTTCGG | CGGGCAGCCTGTGAGACCTTTGA |
| SYBR green incorporation: | |||
| TNF-α | CCGATGGGTTGTACCTTGTC | GTGGGTGAGGAGCACGTAGT | |
| TNFR1 | GACCGGGAGAAGAGGGATAG | GTTCCTTTGTGGCACTTGGT | |
| AQP4 | AGGTGCCCTTTATGAGTATG | CTTCCCTTCTTCTCTTCTCC | |
| GAPDH | GGCAAATTCAACGGCACAGT | AGATGGTGATGGGCTTCCC | |
Acknowledgments
This work was supported by National Institutes of Health Grants R01DK41873,R01DK55357, and R01AI43579
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Alexander JJ, Bao L, Jacob A, Kraus DM, Holers VM, Quigg RJ. Administration of the soluble complement inhibitor, Crry-Ig, reduces inflammation and aquaporin 4 expression in lupus cerebritis. Biochim Biophys Acta. 2003;1639:169–176. doi: 10.1016/j.bbadis.2003.09.005. [DOI] [PubMed] [Google Scholar]
- Alexander JJ, Jacob A, Bao L, MacDonald RL, Quigg RJ. Complement-Dependent Apoptosis and Inflammatory Gene Changes in Murine Lupus Cerebritis. J Immunol. 2005;175:8312–8319. doi: 10.4049/jimmunol.175.12.8312. [DOI] [PubMed] [Google Scholar]
- Alexander JJ, Jacob A, Vezina P, Sekine H, Gilkeson GS, Quigg RJ. Absence of functional alternative complement pathway alleviates lupus cerebritis. Eur J Immunol. 2007;37:1691–1701. doi: 10.1002/eji.200636638. [DOI] [PubMed] [Google Scholar]
- Allan SM, Rothwell NJ. Cytokines and acute neurodegeneration. Nat Rev Neurosci. 2001;2:734–744. doi: 10.1038/35094583. [DOI] [PubMed] [Google Scholar]
- Amrani Y, Lazaar AL, Hoffman R, Amin K, Ousmer S, Panettieri RA., Jr Activation of p55 tumor necrosis factor-alpha receptor-1 coupled to tumor necrosis factor receptor-associated factor 2 stimulates intercellular adhesion molecule-1 expression by modulating a thapsigargin-sensitive pathway in human tracheal smooth muscle cells. Mol Pharmacol. 2000;58:237–245. doi: 10.1124/mol.58.1.237. [DOI] [PubMed] [Google Scholar]
- Askalan R, Deveber G, Ho M, Ma J, Hawkins C. Astrocytic-inducible nitric oxide synthase in the ischemic developing human brain. Pediatric Research. 2006;60:687–692. doi: 10.1203/01.pdr.0000246226.89215.a6. [DOI] [PubMed] [Google Scholar]
- Badaut J, Lasbennes F, Magistretti PJ, Regli L. Aquaporins in brain: distribution, physiology, and pathophysiology. J Cereb Blood Flow Metab. 2002;22:367–378. doi: 10.1097/00004647-200204000-00001. [DOI] [PubMed] [Google Scholar]
- Bi XL, Yang JY, Dong YX, Wang JM, Cui YH, Ikeshima T, Zhao YQ, Wu CF. Resveratrol inhibits nitric oxide and TNF-alpha production by lipopolysaccharide-activated microglia. Int Immunopharmacol. 2005;5:185–193. doi: 10.1016/j.intimp.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Brown GC, Bal-Price A. Inflammatory neurodegeneration mediated by nitric oxide, glutamate, and mitochondria. Mol Neurobiol. 2003;27:325–355. doi: 10.1385/MN:27:3:325. [DOI] [PubMed] [Google Scholar]
- Choi DK, Lee H, Jeong J, Lim B, Suk K. Differential effects of ethanol on glial signal transduction initiated by lipopolysaccharide and interferon-gamma. J Neurosci Res. 2005;82:225–231. doi: 10.1002/jnr.20647. [DOI] [PubMed] [Google Scholar]
- Cunningham PN, Dyanov HM, Park P, Wang J, Newell KA, Quigg RJ. Acute renal failure in endotoxemia is caused by TNF acting directly on TNF receptor-1 in kidney. J Immunol. 2002;168:5817–5823. doi: 10.4049/jimmunol.168.11.5817. [DOI] [PubMed] [Google Scholar]
- Davies DC. Blood-brain barrier breakdown in septic encephalopathy and brain tumours. J Anat. 2002;200:639–646. doi: 10.1046/j.1469-7580.2002.00065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davson H, Segal MB. Physiology of the CSF and the blood-brain barrier. New York: CRC; 1995. [Google Scholar]
- Davson H, Zlokovic BV, Rakic L, Segal MB. An introduction to the blood-brain barrier. London: Macmillan; 1993. [Google Scholar]
- Dong Y, Benveniste EN. Immune function of astrocytes. Glia. 2001;36:180–190. doi: 10.1002/glia.1107. [DOI] [PubMed] [Google Scholar]
- Dopp JM, kenzie-Graham A, Otero GC, Merrill JE. Differential expression, cytokine modulation, and specific functions of type-1 and type-2 tumor necrosis factor receptors in rat glia. J Neuroimmunol. 1997;75:104–112. doi: 10.1016/s0165-5728(97)00009-x. [DOI] [PubMed] [Google Scholar]
- Eng LF, Ghirnikar RS. GFAP and astrogliosis. Brain Pathol. 1994;4:229–237. doi: 10.1111/j.1750-3639.1994.tb00838.x. [DOI] [PubMed] [Google Scholar]
- Eng LF, Ghirnikar RS, Lee YL. Glial fibrillary acidic protein: GFAP-thirty-one years (1969-2000) Neurochem Res. 2000;25:1439–1451. doi: 10.1023/a:1007677003387. [DOI] [PubMed] [Google Scholar]
- Eng LF, Yu AC, Lee YL. Astrocytic response to injury. Prog Brain Res. 1992;94:353–365. doi: 10.1016/s0079-6123(08)61764-1. [DOI] [PubMed] [Google Scholar]
- Enkhbaatar P, Cox R, Traber LD, Maybauer MO, Maybauer DM, Nakano YY, Hawkins H, Schmalstieg F, Herndon D, Traber D. Role of inducible nitric oxide synthase in septic shock. Faseb Journal. 2006;20:A1391. doi: 10.1097/01.shk.0000209525.50990.28. [DOI] [PubMed] [Google Scholar]
- Gahm C, Holmin S, Wiklund PN, Brundin L, Mathiesen T. Neuroprotection by selective inhibition of inducible nitric oxide synthase after experimental brain contusion. J Neurotrauma. 2006;23:1343–1354. doi: 10.1089/neu.2006.23.1343. [DOI] [PubMed] [Google Scholar]
- Gardenfors A, Nilsson F, Skagerberg G, Ungerstedt U, Nordstrom CH. Cerebral physiological and biochemical changes during vasogenic brain oedema induced by intrathecal injection of bacterial lipopolysaccharides in piglets. Acta Neurochir (Wien) 2002;144:601–608. doi: 10.1007/s00701-002-0954-1. [DOI] [PubMed] [Google Scholar]
- Gimenez MA, Sim JE, Russell JH. TNFR1-dependent VCAM-1 expression by astrocytes exposes the CNS to destructive inflammation. J Neuroimmunol. 2004b;151:116–125. doi: 10.1016/j.jneuroim.2004.02.012. [DOI] [PubMed] [Google Scholar]
- Gimenez MA, Sim JE, Russell JH. TNFR1-dependent VCAM-1 expression by astrocytes exposes the CNS to destructive inflammation. J Neuroimmunol. 2004a;151:116–125. doi: 10.1016/j.jneuroim.2004.02.012. [DOI] [PubMed] [Google Scholar]
- Granert C, Raud J, Xie X, Lindquist L, Lindbom L. Inhibition of leukocyte rolling with polysaccharide fucoidin prevents pleocytosis in experimental meningitis in the rabbit. J Clin Invest. 1994;93:929–936. doi: 10.1172/JCI117098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green R, Scott LK, Minagar A, Conrad S. Sepsis associated encephalopathy (SAE): a review. Front Biosci. 2004;9:1637–1641. doi: 10.2741/1250. [DOI] [PubMed] [Google Scholar]
- Gurney KJ, Estrada EY, Rosenberg GA. Blood-brain barrier disruption by stromelysin-1 facilitates neutrophil infiltration in neuroinflammation. Neurobiol Dis. 2006;23:87–96. doi: 10.1016/j.nbd.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Hawkins RD, Son H, Arancio O. Nitric oxide as a retrograde messenger during long-term potentiation in hippocampus. Nitric Oxide in Brain Development, Plasticity and Disease. 1998;118:155–172. doi: 10.1016/s0079-6123(08)63206-9. [DOI] [PubMed] [Google Scholar]
- Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- Henninger DD, Panes J, Eppihimer M, Russell J, Gerritsen M, Anderson DC, Granger DN. Cytokine-induced VCAM-1 and ICAM-1 expression in different organs of the mouse. J Immunol. 1997a;158:1825–1832. [PubMed] [Google Scholar]
- Henninger DD, Panes J, Eppihimer M, Russell J, Gerritsen M, Anderson DC, Granger DN. Cytokine-induced VCAM-1 and ICAM-1 expression in different organs of the mouse. J Immunol. 1997b;158:1825–1832. [PubMed] [Google Scholar]
- Intiso D, Zarrelli MM, Lagioia G, Di RF, Checchia De AC, Simone P, Tonali P, Cioffi Dagger RP. Tumor necrosis factor alpha serum levels and inflammatory response in acute ischemic stroke patients. Neurol Sci. 2004;24:390–396. doi: 10.1007/s10072-003-0194-z. [DOI] [PubMed] [Google Scholar]
- Jellinger KA, Stadelmann C. Mechanisms of cell death in neurodegenerative disorders. J Neural Transm Suppl. 2000a;59:95–114. doi: 10.1007/978-3-7091-6781-6_13. [DOI] [PubMed] [Google Scholar]
- Jellinger KA, Stadelmann CH. The enigma of cell death in neurodegenerative disorders. J Neural Transm Suppl. 2000b:21–36. doi: 10.1007/978-3-7091-6301-6_2. [DOI] [PubMed] [Google Scholar]
- Kubes P, Kanwar S, Niu XF, Gaboury JP. Nitric oxide synthesis inhibition induces leukocyte adhesion via superoxide and mast cells. FASEB J. 1993;7:1293–1299. doi: 10.1096/fasebj.7.13.8405815. [DOI] [PubMed] [Google Scholar]
- Lee SC, Dickson DW, Brosnan CF. Interleukin-1, nitric oxide and reactive astrocytes. Brain Behav Immun. 1995;9:345–354. doi: 10.1006/brbi.1995.1032. [DOI] [PubMed] [Google Scholar]
- Lee SM, Yune TY, Kim SJ, Kim YC, Oh YJ, Markelonis GJ, Oh TH. Minocycline inhibits apoptotic cell death via attenuation of TNF-alpha expression following iNOS/NO induction by lipopolysaccharide in neuron/glia co-cultures. J Neurochem. 2004;91:568–578. doi: 10.1111/j.1471-4159.2004.02780.x. [DOI] [PubMed] [Google Scholar]
- Loddick SA, Rothwell NJ. Mechanisms of tumor necrosis factor alpha action on neurodegeneration: interaction with insulin-like growth factor-1. Proc Natl Acad Sci U S A. 1999;96:9449–9451. doi: 10.1073/pnas.96.17.9449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas R, Lou J, Morel DR, Ricou B, Suter PM, Grau GE. TNF receptors in the microvascular pathology of acute respiratory distress syndrome and cerebral malaria. J Leukoc Biol. 1997;61:551–558. doi: 10.1002/jlb.61.5.551. [DOI] [PubMed] [Google Scholar]
- Manley GT, Binder DK, Papadopoulos MC, Verkman AS. New insights into water transport and edema in the central nervous system from phenotype analysis of aquaporin-4 null mice. Neuroscience. 2004;129:983–991. doi: 10.1016/j.neuroscience.2004.06.088. [DOI] [PubMed] [Google Scholar]
- Marcus JS, Karackattu SL, Fleegal MA, Sumners C. Cytokine-stimulated inducible nitric oxide synthase expression in astroglia: role of Erk mitogen-activated protein kinase and NF-kappaB. Glia. 2003;41:152–160. doi: 10.1002/glia.10168. [DOI] [PubMed] [Google Scholar]
- Marshall D, Dangerfield JP, Bhatia VK, Larbi KY, Nourshargh S, Haskard DO. MRL/lpr lupus-prone mice show exaggerated ICAM-1-dependent leucocyte adhesion and transendothelial migration in response to TNF-alpha. Rheumatology (Oxford) 2003;42:929–934. doi: 10.1093/rheumatology/keg251. [DOI] [PubMed] [Google Scholar]
- Merrill JE, Benveniste EN. Cytokines in inflammatory brain lesions: helpful and harmful. Trends Neurosci. 1996;19:331–338. doi: 10.1016/0166-2236(96)10047-3. [DOI] [PubMed] [Google Scholar]
- Nadeau S, Rivest S. Effects of circulating tumor necrosis factor on the neuronal activity and expression of the genes encoding the tumor necrosis factor receptors (p55 and p75) in the rat brain: a view from the blood-brain barrier. Neuroscience. 1999;93:1449–1464. doi: 10.1016/s0306-4522(99)00225-0. [DOI] [PubMed] [Google Scholar]
- Norenberg MD. Astrocytes in neuronal degeneration. Journal of Neurochemistry. 1996;66:S79. [Google Scholar]
- Norenberg MD. Role of astrocytes in neurologic disease: An introduction. Journal of Neurochemistry. 1998;70:S75. [Google Scholar]
- Norenberg MD, Rao KV, Jayakumar AR. Mechanisms of ammonia-induced astrocyte swelling. Metab Brain Dis. 2005;20:303–318. doi: 10.1007/s11011-005-7911-7. [DOI] [PubMed] [Google Scholar]
- Pan W, Banks WA, Kastin AJ. Blood-brain barrier permeability to ebiratide and TNF in acute spinal cord injury. Exp Neurol. 1997a;146:367–373. doi: 10.1006/exnr.1997.6533. [DOI] [PubMed] [Google Scholar]
- Pan W, Banks WA, Kastin AJ. Blood-brain barrier permeability to ebiratide and TNF in acute spinal cord injury. Exp Neurol. 1997b;146:367–373. doi: 10.1006/exnr.1997.6533. [DOI] [PubMed] [Google Scholar]
- Papadopoulos MC, Davies DC, Moss RF, Tighe D, Bennett ED. Pathophysiology of septic encephalopathy: a review. Crit Care Med. 2000;28:3019–3024. doi: 10.1097/00003246-200008000-00057. [DOI] [PubMed] [Google Scholar]
- Radzivil GG, Beloborodov VB, Bronikin I. Hemodynamics and rheologic properties of the blood in meningococcemia associated with meningitis and complicated by septic shock and intracranial hypertension. Anesteziol Reanimatol. 1990:28–33. [PubMed] [Google Scholar]
- Rao KVR, Chen M, Simard JM, Norenberg MD. Increased aquaporin-4 expression in ammonia-treated cultured astrocytes. Neuroreport. 2003;14:2379–2382. doi: 10.1097/00001756-200312190-00018. [DOI] [PubMed] [Google Scholar]
- Romero LI, Tatro JB, Field JA, Reichlin S. Roles of IL-1 and TNF-alpha in endotoxin-induced activation of nitric oxide synthase in cultured rat brain cells. Am J Physiol. 1996;270:R326–R332. doi: 10.1152/ajpregu.1996.270.2.R326. [DOI] [PubMed] [Google Scholar]
- Schafers M, Schmidt C, Vogel C, Toyka KV, Sommer C. Tumor necrosis factor-alpha (TNF) regulates the expression of ICAM-1 predominantly through TNF receptor 1 after chronic constriction injury of mouse sciatic nerve. Acta Neuropathol (Berl) 2002;104:197–205. doi: 10.1007/s00401-002-0541-9. [DOI] [PubMed] [Google Scholar]
- Tsao N, Hsu HP, Wu CM, Liu CC, Lei HY. Tumour necrosis factor-alpha causes an increase in blood-brain barrier permeability during sepsis. J Med Microbiol. 2001;50:812–821. doi: 10.1099/0022-1317-50-9-812. [DOI] [PubMed] [Google Scholar]
- Vizuete ML, Venero JL, Vargas C, Ilundain AA, Echevarria M, Machado A, Cano J. Differential upregulation of aquaporin-4 mRNA expression in reactive astrocytes after brain injury: potential role in brain edema. Neurobiol Dis. 1999;6:245–258. doi: 10.1006/nbdi.1999.0246. [DOI] [PubMed] [Google Scholar]
- Wispelwey B, Hansen EJ, Scheld WM. Haemophilus influenzae outer membrane vesicle-induced blood-brain barrier permeability during experimental meningitis. Infect Immun. 1989;57:2559–2562. doi: 10.1128/iai.57.8.2559-2562.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Froio RM, Sciuto TE, Dvorak AM, Alon R, Luscinskas FW. ICAM-1 regulates neutrophil adhesion and transcellular migration of TNF-alpha-activated vascular endothelium under flow. Blood. 2005;106:584–592. doi: 10.1182/blood-2004-12-4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin L, Ohtaki H, Nakamachi T, Kudo Y, Makino R, Shioda S. Delayed expressed TNFR1 co-localize with ICAM-1 in astrocyte in mice brain after transient focal ischemia. Neurosci Lett. 2004a;370:30–35. doi: 10.1016/j.neulet.2004.07.083. [DOI] [PubMed] [Google Scholar]
- Yin L, Ohtaki H, Nakamachi T, Kudo Y, Makino R, Shioda S. Delayed expressed TNFR1 co-localize with ICAM-1 in astrocyte in mice brain after transient focal ischemia. Neurosci Lett. 2004b;370:30–35. doi: 10.1016/j.neulet.2004.07.083. [DOI] [PubMed] [Google Scholar]
- Yuan J, Yankner BA. Apoptosis in the nervous system. Nature. 2000;407:802–809. doi: 10.1038/35037739. [DOI] [PubMed] [Google Scholar]
- Yung HW, Bal-Price AK, Brown GC, Tolkovsky AM. Nitric oxide-induced cell death of cerebrocortical murine astrocytes is mediated through p53- and Bax-dependent pathways. J Neurochem. 2004;89:812–821. doi: 10.1111/j.1471-4159.2004.02395.x. [DOI] [PubMed] [Google Scholar]