

Genome-wide association studies have identified several single nucleotide polymorphisms (SNPs) as reproducibly associated with risk of myocardial infarction (MI)1-3, a leading cause of death and disability. We tested both SNPs and copy number variants (CNVs) for association with early-onset MI in a large sample of 2,967 cases of early-onset MI and 3,075 matched controls. The design called for any variant with P < 0.001 to be tested for replication in up to 18,822 additional individuals. SNPs at eight loci reached genome-wide significance, two of which are newly identified: PHACTR1 (P = 6 × 10-10) and MRPS6/KCNE2 (P = 2 × 10-9). We tested 554 common CNVs (> 1% frequency) for association with MI; none met the pre-specified threshold for replication testing (P < 10-3), and the Q-Q plot did not deviate from the null distribution. We identified 8,065 rare CNVs but did not detect a greater CNV burden in cases as compared to controls, in genes as compared to the genome as a whole, or at any individual locus. Common SNPs at eight loci were reproducibly associated with risk of MI but a systematic well-powered test of common and rare CNVs failed to identify additional associations to risk of MI.
Myocardial infarction (MI) is heritable4 and among the leading causes of death and disability worldwide5. Whereas the majority of MIs occur in individuals >65 years old, 5-10% of new MIs occur in younger patients and these events are associated with substantially greater heritability5,6. Thus, early-onset MI is a promising phenotype for genetic mapping.
Genome-wide association studies (GWASs) of common SNPs have been reported for MI and coronary artery disease1-3,7, with each study finding common SNPs on chromosome 9p21.3 associated with MI or coronary artery disease. In addition to 9p21.3, these papers proposed at least eight other loci as harboring SNPs associated with coronary artery disease. Some of these loci await definitive replication, but even if all were valid they would explain a small fraction of the risk of MI.
Structural variants, another class of human DNA sequence variation, may account for some of the unexplained heritability in MI and other common diseases8,9. Common CNVs have been associated with Crohn’s disease10 and body mass index11 and rare CNVs have been related to risk for autism12 and schizophrenia13-16. To our knowledge, no integrated assessment of SNPs and CNVs in the same samples has been reported for MI or any other trait. Several technological developments make such systematic surveys now possible including hybrid oligonucleotide microarrays17 and analytical methods18 to simultaneously assess SNPs and CNVs genome-wide in each sample.
We designed a three-staged GWAS of early-onset MI with SNPs, common CNVs, and rare CNVs (Figure 1). Stage 1 consisted of the Myocardial Infarction Genetics Consortium (MIGen), a collection of 2,967 cases of early-onset MI (in men ≤50 years old or women ≤60 years old) and 3,075 age- and sex-matched controls free of MI from six international sites: Boston and Seattle in the United States as well as Sweden, Finland, Spain, and Italy (Table 1 and Supplementary Methods). The mean age at the time of MI was 41 years among males and 47 years among females.
Figure 1. Study Design.

The genome-wide association study consisted of three stages with an evaluation of common single nucleotide polymorphisms, common copy number variants, and rare copy number variants in Stage 1. The design called for all variants with a P < 0.001 to be taken forward to Stage 2. As only SNPs met this criterion, 1441 SNPs were taken forward to Stage 2. A total of 33 SNPs were tested in Stage 3. Statistical evidence for association was combined across Stages 1, 2, and 3 using meta-analysis.
Table 1. Participant characteristics of case and control subjects in Stage 1 of the genome-wide association screen.
| Study | Italian ATVB Study | Heart Attack Risk in Puget Sound | REGICOR | MGH Premature Coronary Artery Disease Study | FINRISK | Malmö Diet and Cancer Study | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||
| cases | controls | cases | controls | cases | controls | cases | controls | cases | controls | cases | controls | |
| N | 1,693 | 1,668 | 505 | 559 | 312 | 317 | 204 | 260 | 167 | 172 | 86 | 99 |
|
Ascertainment scheme |
hospital- based |
hospital- based |
community- based |
community- based |
hospital- based |
drawn from community-based cohort | hospital- based |
hospital- based |
drawn from population-based cohort | nested case-cohort | drawn from population-based cohort | nested case-cohort |
|
MI age criterion |
men or women ≤ 45 | -- | men ≤50 or women ≤ 60 | -- | men ≤50 or women ≤ 60 | -- | men ≤50 or women ≤ 60 | -- | men ≤50 or women ≤ 60 | -- | men ≤50 or women ≤ 60 | -- |
| Country of origin * | Italy | Italy | U.S. | U.S. | Spain | Spain | U.S. | U.S. | Finland | Finland | Sweden | Sweden |
| Mean age (y) † | 39.4 ± 4.9 | 39.3 ± 5.0 | 46.0 ± 6.9 | 45.2 ± 7.3 | 45.9 ± 5.8 | 46.0 ± 5.6 | 47.0 ± 6.1 | 53.8 ± 11.1 | 47.1 ± 6.2 | 47.1 ± 6.0 | 48.5 ± 4.4 | 48.7 ± 4.6 |
|
Female gender (%) |
11.4 | 11.6 | 51.1 | 55.5 | 20.2 | 21.5 | 29.9 | 33.5 | 33.5 | 31.4 | 41.9 | 42.4 |
| Ever cigarette smoking (%) | 87.0 | 49.3 | 73.9 | 41.7 | 82.8 | 61.9 | 74.9 | 57.3 | 74.4 | 58.2 | 87.2 | 61.6 |
|
Hypertension (%) ‡ |
32.6 | 11.9 | 50.5 | 30.8 | 38.0 | 31.5 | 33.5 | 25.3 | 72.5 | 68.0 | 81.4 | 62.6 |
| Diabetes mellitus (%) § | 7.8 | 0.8 | 14.9 | 3.0 | 14.8 | 6.1 | 19.2 | 0.4 | 17.7 | 5.9 | 4.7 | 1.0 |
|
Hypercholest erolemia (%) ¶ |
60.4 | 44.4 | 43.7 | 26.0 | 48.9 | 33.1 | 79.0 | 31.3 | 75.2 | 48.2 | 37.2 | 1.0 |
| Body mass index (kg/m2) | 26.7 ± 4.2 | 25.0 ± 3.3 | 29.2 ± 6.8 | 26.9 ± 5.7 | 27.5 ± 4.2 | 27.0 ± 3.9 | 30.0 ± 7.0 | 27.9 ± 6.5 | 29.6 ± 5.0 | 27.7 ± 4.0 | 26.9 ± 4.2 | 25.7 ± 4.3 |
Values with ‘±’ are means ± s.d. The body-mass index is the weight in kilograms divided by the square of the height in meters.
All cases and controls were of European ancestry.
Mean age at MI for cases and at age of recruitment for controls
Hypertension was defined as a previous diagnosis of hypertension, on anti-hypertensive therapy, or with recorded systolic blood pressure ≥ 140 mmHg or diastolic blood pressure ≥ 90 mmgHg.
Diabetes mellitus was defined as a previous diagnosis of diabetes or treatment with anti-diabetic medications.
Hypercholesterolemia was defined as a previous diagnosis of hypercholesterolemia or treatment with lipid-lowering therapy
Variants with P < 0.001 were advanced through two stages of replication (Figure 1, see Methods for power calculations). In total, 1,441 SNPs, including a SNP at each of eight loci recently proposed from GWA or candidate gene studies for coronary artery disease3,7,19, were taken forward into Stage 2, an in silico analysis of these SNPs in four recently completed GWA studies for MI. Stage 2 consisted of an effective symmetric sample size of 3,942 cases of MI and 3,942 controls (Supplementary Methods and Supplementary Table 1). Thirty-three SNPs were taken forward from Stage 2 into Stage 3, consisting of an additional 6 studies with an effective symmetric sample size of 5,469 cases of MI and 5,469 controls (Supplementary Methods and Supplementary Table 2). Stage 3 included 25 SNPs with the best combined statistical evidence in Stages 1 and 2 and 8 SNPs from previously reported loci (Methods).
After Stages 1, 2, and 3, we observed that SNPs at 8 loci were associated with MI at a pre-specified threshold for genome-wide significance of P < 5 × 10-8 (corresponding to P < 0.05 after adjusting for ~ 1 million independent tests20) (Table 2). Six of the eight previously-reported associations were confirmed (Table 2) with P ranging from 2 × 10-8 to 1 × 10-41. As the Stage 2 samples were used to implicate some of these previous findings, the data we present are not fully independent of prior reports. These six genetic association signals map to 9p21.3, CXCL12, CELSR2/PSRC1/SORT1, MIA3, LDLR and PCSK93,7. Three of the SNPs (those at the CELSR2/PSRC1/SORT1, LDLR, and PCSK9 loci) have been also previously shown to relate to plasma low-density lipoprotein cholesterol, a causal risk factor for MI7,21. The risk alleles at the eight loci ranged in frequency from 13% to 84%. Each copy of the risk allele conferred excess odds of MI ranging from 13% to 28%.
Table 2. Single nucleotide polymorphisms associated with risk for early-onset myocardial infarction.
| Stage 1 (MIGen) | Stage 2 (WTCCC, GerMIFSI, PennCATH, Medstar) | Stage 3 (AMI Gene, Verona, MAHI, IFS, GerMIFSII, INTERHEART) | Combined Stage 1 + 2 + 3 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|||||||||||||||||
| SNP | Chr | position NCBI35 (bp) |
non-risk allele |
risk allele |
risk allele frequency |
gene(s) of interest in associated interval |
OR (95% CI) |
P value [IC] |
OR (95% CI) |
P value |
OR (95% CI) |
P value |
neff | OR (95% CI)† |
P value‡ |
P het | n samples for 80% power§ |
| Novel Loci | |||||||||||||||||
| rs12526453 | 6 | 13,035,530 | G | C | 0.65 | PHACTR1 | 1.15 (1.07-1.24) |
4.60E-04 | 1.12 (1.05-1.21) |
3.64E-04 | 1.12 (1.05-1.19) |
2.31E-04 | 12,434 | 1.13 (1.09-1.17) |
6.54E-10 | 0.07 | 2,581 |
| rs9982601 | 21 | 34,520,998 | C | T | 0.13 |
SLC5A3/ MRPS6/ KCNE2 |
1.20 (1.07-1.33) |
7.82E-04 [0.93] |
1.34 (1.22-1.47) |
1.74E-09 | 1.06 (0.97-1.17) |
0.28 | 11,173 | 1.19 (1.13-1.27) |
2.12E-09 | 0.05 | 2,119 |
| Previously Reported Loci | |||||||||||||||||
| rs4977574 | 9 | 22,088,574 | A | G | 0.56 | CDKN2A/2B | 1.25 (1.16-1.34) |
6.67E-09 | 1.38 (1.29-1.47) |
6.87E-22 | 1.25 (1.18-1.31) |
2.35E-15 | 12,434 | 1.28 (1.24-1.33) |
1.08E-41 | 0.08 | 650 |
| rs1746048 | 10 | 44,095,830 | T | C | 0.84 | CXCL12 | 1.22 (1.10-1.34) |
1.61E-04 | 1.28 (1.16-1.42) |
1.75E-06 | 1.12 (1.04-1.21) |
3.35E-03 | 12,434 | 1.19 (1.13-1.25) |
8.14E-11 | 0.17 | 2,521 |
| rs646776 | 1 | 109,530,572 | C | T | 0.81 |
CELSR2/ PSRC1/ SORT1 |
1.11 (1.02-1.22) |
0.04 [0.99] |
1.36 (1.21-1.53) |
3.91E-07 | 1.16 (1.08-1.25) |
1.64E-05 | 9,364 | 1.18 (1.12-1.25) |
9.36E-11 | 3.7E-03 | 2,366 |
| rs17465637 | 1 | 220,890,152 | A | C | 0.72 | MIA3 | 1.18 (1.08-1.27) |
1.50E-04 | 1.19 (1.08-1.31) |
4.94E-04 | 1.09 (1.03-1.16) |
4.48E-03 | 10,961 | 1.13 (1.09-1.19) |
1.33E-08 | 8.0E-03 | 2,982 |
| rs1122608 | 19 | 11,024,601 | T | G | 0.75 | LDLR | 1.18 (1.09-1.28) |
1.72E-04 | 1.18 (1.10-1.28) |
2.60E-05 | 1.07 (1.00-1.15) |
0.041 | 11,562 | 1.14 (1.09-1.19) |
1.49E-08 | 7.2E-04 | 2,858 |
| rs11206510 | 1 | 55,268,627 | C | T | 0.81 | PCSK9 | 1.12 (1.02-1.23) |
0.02 | 1.16 (1.07-1.26) |
9.07E-04 | 1.18 (1.09-1.27) |
8.36E-05 | 12,219 | 1.15 (1.10-1.21) |
2.02E-08 | 0.19 | 3,174 |
| Loci Requiring Additional Followup | |||||||||||||||||
| rs6725887* | 2 | 203,454,130 | T | C | 0.14 | WDR12 | 1.24 (1.12-1.38) |
8.55E-05 | 1.15 (1.04-1.26) |
2.99E-03 | 1.11 (1.02-1.21) |
0.029 | 12,434 | 1.16 (1.10-1.22) |
4.29E-07 | 0.19 | 2,753 |
| rs7947046 | 11 | 61,016,327 | C | T | 0.92 | SYT7 | 1.29 (1.13-1.47) |
3.40E-04 | 1.24 (1.09-1.41) |
2.16E-03 | 1.16 (1.03-1.31) |
0.032 | 10,414 | 1.22 (1.14-1.32) |
4.31E-07 | 1.8E-03 | 2,588 |
|
| |||||||||||||||||
| Maximum available effective sample size | 6,046 | 7,844 | 10,938 | 24,828 | |||||||||||||
UNDERLINE denotes imputed SNP in Stage 1; BOLD denotes minor allele
For all studies except INTERHEART, where rs4675310 was substituted as a close to perfect proxy to rs6725887 [Hapmap CEU r2 = 1.0]
Odds ratio based on a fixed-effect based meta-analysis of odds ratios.
P-value based on a weighted z-score meta-analysis
Symmetric case/control sample size assuming a type-I error rate of 0.05, the combined Stage 1 + 2 + 3 estimate of the OR, risk allele frequency from Stage I, an additive genetic model, and a prevalence of 3%
Three of the loci previously suggested by Samani et al.3 did not meet our pre-specified threshold of P < 5 × 10-8. Across Stages 1, 2, and 3, the statistical evidence was the following: rs17228212 in SMAD3 (odds ratio 1.03, 95% confidence interval 0.99 - 1.07, P = 0.15); rs2943634 on 2q36 (odds ratio 1.05, 95% confidence interval 1.01 - 1.10, P = 0.01); and rs6922269 in MTHFD1L (odds ratio 1.09, 95% confidence interval 1.05 - 1.14, P = 2 × 10-5).
Two novel associations were observed with genome-wide significance: (i) in an intron of phophastase and actin regulator 1 (PHACTR1) on chromosome 6 (rs12526453, odds ratio 1.13, P = 7 × 10-10) and (ii) in an intergenic region between mitochondrial ribosomal protein S6 (MRPS6), solute carrier family 5 (inositol transporters) member 3 (SLC5A3) and potassium voltage-gated channel, Isk-related family, member 2 (KCNE2) on chromosome 21 (rs9982601, odds ratio 1.19, P = 2 × 10-9). PHACTR1 is an inhibitor of protein phosphatase 1, an enzyme that dephosphorylates serine and threonine residues on a range of proteins22. MRPS6 encodes a subunit of the mitochondrial ribosomal protein 28S23. SLC5A3 is a gene embedded within MRPS6 and encodes a protein that transports sodium and myo-inositol in response to hypertonic stress24. KCNE2 encodes a subunit of a potassium channel and mutations in this gene cause inherited arrhythmias25. The mechanisms by which gene(s) at these two loci lead to MI remain to be defined.
At two additional new loci (in an intron of WDR12 and near SYT7), the statistical evidence for association across Stages 1, 2, and 3 was consistent (combined P for each at 4 × 10-7) but did not meet our pre-specified genome-wide threshold (Table 2). These loci require follow-up in additional samples.
Of the eight validated loci, non-coding SNPs at 9p21.3 have been the most widely replicated, confer the largest effect size and are supported by the strongest statistical evidence26. While it is possible that 9p21.3 SNPs act through as-yet unidentified coding variants, non-coding SNPs may affect function by altering level of gene expression. Thus, we explored whether the 9p21.3 SNP from our study might be related to mRNA level of nearby genes in three biologically-relevant human tissues - liver, subcutaneous fat, and visceral fat (Methods).
The MI-associated SNP at 9p21.3 (rs4977574) was strongly associated with mRNA level of cyclin-dependent kinase inhibitor 2B (CDKN2B), a gene located ~89 kilobases from the SNP. Compared with the mRNA level in a reference pool of individuals, carriers of the risk G allele at 9p21.3 had about the same level of expression of CDKN2B in subcutaneous fat tissue whereas carriers of the non-risk A allele had ~15% lower transcript level (P = 4 × 10-6 in 698 subcutaneous fat samples, Figure 2). The same SNP was also associated with CDKN2B transcript level in visceral fat tissue (P = 1 × 10-4) but not associated in human liver (P = 0.84). In each of the three tissues, this genotype was not associated with mRNA level of other neighboring transcripts on 9p21.3 including CDKN2A, MTAP, or ANRIL (P > 0.05 for each genotype-transcript association). CDKN2B, a downstream target of the transforming growth factor beta pathway, has been shown to decrease cell survival27. These results suggest the hypothesis that genetic variation at 9p21.3 leads to atherosclerosis through CDKN2B.
Figure 2. CDKN2B messenger RNA expression in subcutaneous fat tissue stratified by rs4977574 genotyped on 9p21.3.

The CDKN2B transcript level in each of 848 subcutaneous fat samples was compared with the mean level in a control mRNA pool of 100 randomly-selected samples. A ratio of the sample transcript level over that in control pool was first calculated and then log-transformed. This percent change is shown on the y-axis with the genotype at rs4977574 shown on the x-axis. Note that the G allele represents the risk allele for MI with each copy of the G allele increasing risk for MI by 28%.
To evaluate the cumulative effect of these eight SNPs on risk for MI, we constructed an MI genotype score comprised of the 8 SNPs, modeling the number of risk alleles carried by each individual in the MIGen GWAS (Stage 1). In logistic regression models including age, gender, and principal components of ancestry, individuals in the top quintile of MI genotype score had a two-fold increased risk for MI compared with bottom quintile (odds ratio 2.05, 95% confidence interval 1.74 to 2.42; P = 4 × 10-25, Table 3). The MI genotype score confers risk of a magnitude comparable to other established risk factors such as plasma low-density lipoprotein cholesterol (odd ratio 1.62, 95% confidence interval 1.17 - 2.25 for top versus bottom quintile as previously reported28).
Table 3. Quintiles of allelic dosage score comprised of eight validated MI single nucleotide polymorphisms and risk for early-onset myocardial infarction.
| Quintile of MI Genotype Score | Odds ratio | 95% confidence interval |
|---|---|---|
| Quintile 1 | 1.0 (reference group) | |
| Quintile 2 | 1.10 | 0.93 - 1.29 |
| Quintile 3 | 1.41 | 1.20 - 1.65 |
| Quintile 4 | 1.60 | 1.36 - 1.89 |
| Quintile 5 | 2.05 | 1.74 - 2.42 |
| P for association of MI genotype score with early-onset MI: 4 × 10-25 | ||
The eight validated MI polymorphisms are as shown in Table 2 and include PHACTR1 rs12526453, SLC5A3/MRPS6/KCNE2 rs9982601, 9p21.3 rs4977574, CXCL12 rs1746048, CELSR2/PSRC1/SORT1 rs646776, MIA3 rs17465637, LDLRs rs1122608, and PCKS9 rs11206510. Risk of early-onset MI was assessed in the 2,967 cases and 3,075 controls from Stage 1.
While the GWA approach has met with some success in MI, these variants, in sum, explain a small fraction of the variance; the current MI genotype score explains only 2.4% of the variance in risk for early-onset MI. Thus, we tested the hypothesis that systematic assessment of structural variants, common and rare, might identify additional loci contributing to MI.
We first used the CANARY algorithm18 to test 554 commonly segregating CNVs (> 1% frequency) for association with early-onset MI in 2,783 cases and 2,865 controls that passed sample quality control for CNV analysis (Methods). The estimated genomic control lambda for the entire set of CNVs was ~1.23; for 316 CNVs with allele frequency greater than 5%, lambda was ~1.05. We did not observe any CNV with evidence for association surpassing our pre-specified threshold for replication of P < 0.001. In fact, the strongest association (P = 0.002, Supplementary Table 3) did not pass the Bonferroni correction for 554 tests, let alone genome-wide significance for SNPs. A plot of the observed versus expected P value distribution did not show deviation from the null distribution (Figure 3).
Figure 3. Plot of observed versus expected P value distribution for association of 554 common copy number variants with early-onset myocardial infarction.
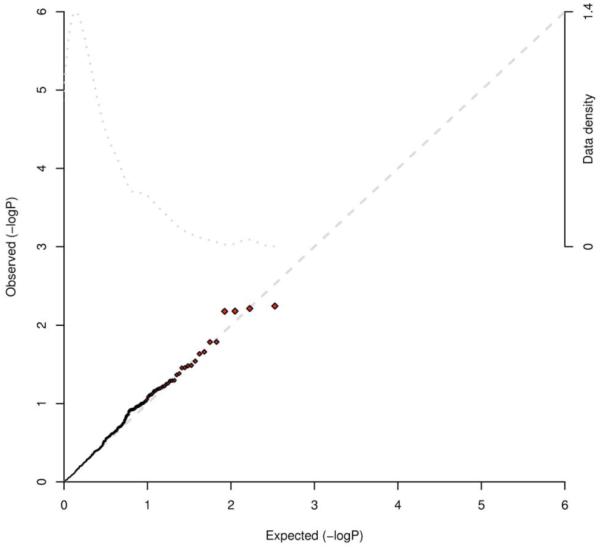
The CANARY algorithm was used to test 554 commonly segregating CNVs (> 1% frequency) for association with early-onset MI in 2,783 cases and 2,865 controls that passed sample quality control for CNV analysis (Methods). The estimated genomic control lambda for the entire set of CNVs was ~1.23; for 316 CNVs with allele frequency greater than 5%, lambda was ~1.05. We did not observe any CNV with evidence for association surpassing our pre-specified threshold for replication of P < 0.001. The observed versus expected P value distribution did not show deviation from the null distribution.
To detect rare CNVs, we used Birdseye18 and restricted analysis to autosomal deletions and duplications that were both rare (< 1% frequency in our samples) and large (greater than 100kb). After stringent quality control filtering (Supplementary Methods), the analysis included 5,955 individuals and 8,065 CNVs (39% deletions). The mean number of rare CNVs per individual was 1.35 and the median was 1.
Using the same methods recently described in a successful study of schizophrenia14, we evaluated case/control differences in rare CNVs across three parameters: the overall burden of rare CNVs genome-wide, the number of genes overlapped by rare CNVs, and the total kilobase extent of rare CNVs. Controlling for sample collection site, there were no case/control differences in genome-wide rare CNV rate (P = 0.39), the number of genes intersected by rare CNVs (P = 0.74) or the total kilobase extent of rare CNVs (P = 0.77). Furthermore, there were no differences in rare CNV rate when restricting analysis to only gene-intersecting rare CNVs (P = 0.55), deletions (P = 0.57) or duplications (P = 0.34). Searching for specific loci with increased rates of rare CNVs in cases versus controls, only 4 regions showed uncorrected P values of P < 0.01; however, the lowest P value after correction for multiple testing was P = 0.96.
In conclusion, we screened common SNPs and CNVs both common and rare for association with early-onset MI in a large sample. Our study suggests four main conclusions. First, there are eight gene regions at which common SNPs are associated with MI with genome-wide significance and replication, two of which were newly implicated by this study. Second, at 9p21.3, we show that the SNP with the best statistical evidence for MI risk is also correlated with expression of a neighboring gene - CDKN2B - in human fat tissue. Third, whereas the effects of the individual SNPs are modest, the overall effect (in a comparison of extreme quintiles) of an eight SNP score (two-fold increased risk for MI) is comparable in predictive value to plasma LDL cholesterol28.
Fourth, and in contrast to the positive results for genetic mapping of MI via SNP analysis, we were unable to detect common or rare CNVs associated with risk for MI. The current analysis is directly comparable to a recent study of schizophrenia that found convincing evidence for rare CNVs associated with disease both at specific loci and for three specific genome-wide burden measures14: both studies are of similar sample size, used the same genotyping platform, and were analyzed by the same methods and by the same analyst. The different results indicate that the genetic architecture of MI may be different than schizophrenia (based on natural selection, genetic complexity or other factors), and that the remaining inherited risk for MI must be due to some combination of common SNPs for which we do not yet have sufficient power, CNVs not measured in our analysis, rare point mutations, and non-additive interactions. However, by systematically measuring all forms of genetic variation in appropriate samples, it should be possible to identify the architecture of each trait and increase information about the pathophysiology of disease.
METHODS
Study design and samples
We conducted a genetic association study with three stages as displayed in Figure 1. Stage 1 consisted of the Myocardial Infarction Genetics Consortium (MIGen), a collection of 2,967 cases of early-onset MI (in men ≤50 years old or women ≤60 years old) and 3,075 age- and sex-matched controls free of MI from six international sites: Boston and Seattle in the United States as well as Sweden, Finland, Spain, and Italy (Table 1). At each site, MI was diagnosed on the basis of autopsy evidence of fatal MI or a combination of chest pain, electrocardiographic evidence of MI, or elevation of one or more cardiac biomarkers (creatine kinase or cardiac troponin). The mean age at the time of MI was 41 years among male cases and 47 years among female cases.
We took forward SNPs into two stages of replication (Stages 2 and 3, Figure 1). 1441 SNPs were tested in Stage 2 based on two criteria: i) strength of statistical evidence in Stage 1 (1433 SNPs from loci with P < 10-3 in Stage 1) or ii) belonging to one of eight reported loci from recent genome-wide association studies for coronary artery disease (a common SNP from each of 9p21.3, near CXCL12, SMAD3, MTHFD1L, MIA3, near CELSR2/PSRC1/SORT1, 2q36, and PCSK9)3,7.
Stage 2 consisted of in silico comparisons with four recently completed GWAS for MI consisting of a symmetric effective sample size of up to 3,942 cases of MI and 3,942 controls. These studies included the Wellcome Trust Case Control Consortium Coronary Heart Disease study3, German MI Family Study I3, PennCATH, and MedStar (Supplementary Table 1). In each Stage 2 study, the analysis was restricted to the phenotype of MI with an age of onset threshold of <66 years for men or women. Although this age cutoff is slightly less restrictive than that used in Stage 1, this cutoff is at or below the mean age of first MI in the US (65 years for men and 70 years for women).
Thirty-three SNPs were taken forward to Stage 3, which consisted of genotyping an additional 6 studies with a symmetric effective sample size of up to 5,469 cases of MI and 5,469 controls. These six studies included Acute MI Gene Study/Dortmund Health Study, Verona Heart Study29, Mid-America Heart Institute Study30, Irish Family Study31, German MI Family Study II, and INTERHEART32 (European ancestry and South Asian ancestry each analyzed separately) (Supplementary Table 2). Stage 3 was comprised of 25 SNPs with the best combined statistical evidence for MI from Stages 1 and 2 (P < 10-5) and the eight previously-reported SNPs discussed above. In each Stage 3 study, the analysis was restricted to the phenotype of MI and in four of the six studies, an age of onset threshold was established at <66 years for men or women.
Genotyping
In Stage 1, we studied 727,496 directly genotyped SNPs (Affymetrix 6.0 GeneChip) that passed quality control filters as described in the Supplementary Appendix. In addition, we used these genotyped SNPs and the phased chromosomes from the HapMap CEU sample to impute genotypes for an additional 1,830,248 SNPs with MACH 1.0 software. In previous work, we have demonstrated that imputation is accurate (average concordance rate of 97.9% between imputed and genotyped data for the same SNP) when using MACH 1.0 in samples of European ancestry with the HapMap CEU phased chromosomes as reference33.
Stage 2 studies were genotyped on either the Affymetrix GeneChip Human Mapping 500K Array Set or Affymetrix 6.0 GeneChip and imputation of HapMap SNPs was performed using either IMPUTE or Mach 1.0 software (Supplementary Table 1).
In Stage 3, genotyping was attempted for 33 SNPs in five studies using the iPLEX MassARRAY platform (Sequenom). In the sixth study, German MI Family Study II, SNPs were genotyped using the Affymetrix 6.0 array.
Association of individual SNP genotypes with MI
In Stage 1, we tested the association of early-onset MI with a combined set of ~2.5 million SNPs (directly genotyped and imputed) using a logistic regression model that accounted for age, gender, and study site. The estimated genomic control λ1000 was low at 1.01, suggesting little residual confounding due to population stratification. Imputed genotypes were tested for association after accounting for uncertainty using the “PROPER” option in the IMPUTE software package.
In addition, we evaluated an alternate method to account for potential confounding by population stratification within samples of European ancestry. We conducted principal component analysis as implemented in PLINK software to define axes of ancestry within the six Stage 1 studies34. The first two principal components separated individuals into clusters that matched study site labels and revealed the well-known north-south cline in allele frequencies across Europe (Supplementary Figure 1). Logistic regression analysis with the first two principal components as covariates (instead of study site) led to nearly identical association results (correlation in association statistics was 0.99). In Stages 2 and 3, within each study, we examined the association of SNPs with MI using logistic regression after adjustment for age and gender.
We used two meta-analytic methods to summarize the statistical evidence for each SNP across Stages 1, 2, and 3. We combined odds ratios for a given reference allele on a logarithmic scale weighted by the inverse of their variances using a fixed-effects model. We also combined evidence for association solely on the basis of P values. For each study, we converted the two-sided P value to a z-statistic and assigned a sign to reflect the direction of the association given the reference allele. Each z-score was then weighted with the squared weights summing to 1 and each sample-specific weight being proportional to the square root of the effective number of individuals in the sample. We summed the weighted z-statistics across studies and converted the summary z-score to a two-sided P value.
Expression quantitative trait analyses
To evaluate whether the 9p21.3 variant also served as an expression quantitative trait locus with putative cis regulatory effects on gene expression traits, we profiled expression levels of 39,280 transcripts and genotyped 782,476 SNPs in 955 human liver samples35. In addition, we evaluated these same transcripts and genotyped 557,240 SNPs in human subcutaneous fat (n=701) and visceral fat samples (n=848). Liver samples were either postmortem or surgical resections from organ donors. The fat samples were collected from subjects undergoing Roux-en-Y surgery between 2000 and 2007. The transcript level in each sample was compared with the mean level in a control mRNA pool of 100 randomly-selected samples. A ratio of the sample transcript level over that in control pool was first calculated and then log transformed. We tested if mean log-ratios differed across 9p21.3 genotype groups using the Kruskal Wallis test.
MI genotype score
We modeled the cumulative number of MI risk alleles carried by each participant in Stage 1. We constructed a score from the eight SNPs exceeding P < 5 × 10-8 in Table 2. The score was composed of allelic dosage (observed counts of 0, 1, or 2 for genotyped SNPs, or fractional allele counts between 0.0 and 2.0 estimated from the imputation procedure for imputed SNPs), weighted by the effect size of that allele on the MI phenotype (to minimize a potential “winner’s curse”, the effect size was drawn from the combined Stage 1 + 2 + 3 evidence), and summed across SNPs. We tested the association of genotype score with MI using logistic regression models after accounting for age, gender, and two principal components of ancestry. We set the lowest quintile of MI genotype score as the referent group and estimated the increase in odds for MI associated with the remaining quintile groups.
Statistical analyses were conducted using either PLINK software or in R.
Common and rare CNV analysis
Utilizing a previously defined copy number polymorphism map based on HapMap, we genotyped a set of polymorphic (greater than 1% sample frequency) autosomal deletion and duplication variants using the CANARY algorithm18. We first conducted quality control filtering at the sample level. We assessed the initial 6,042 samples for quality in copy-number genotyping using three quality metrics reported by the Birdseye method. We measured the average copy number genome estimates reported by the Birdseye Hidden Markov Model18, and we removed any sample which showed excessively high or low average copy number estimates (> 3 standard deviations than the average genome-wide). Second, we measured the variability in SNP and copy number polymorphism probe intensities, with each standardized per chromosome. We removed any sample with excessive variability in these estimates on average genome-wide (> 3 standard deviations than the average genome-wide). Next, we removed any sample where more than 2 chromosomes failed any of these three metrics (> 3 standard deviations in estimated copy number or excessive SNP or CNV variability for chromosome). Finally, for samples that had 1 or 2 chromosomes failing these measures, rather than failing the sample, we treated the data as missing. As a result, 5,648 samples were copy number genotyped with CANARY software18.
We genotyped these samples for the previously defined set of 1,315 copy number polymorphisms characterized on the HapMap sample17. As an initial quality control step, we removed any variant where more than 10% of the copy calls were uncertain (confidence score > 0.1) or missing. In addition, we focused on a set of polymorphisms where at least one allele had a frequency greater than 1%. This restricted our analysis to 614 copy number polymorphic regions. An additional 59 CNVs were removed for inconsistent genotyping. Thus, we focused on a set of 554 copy number variable regions observed to be polymorphic and well genotyped in a set of 2,783 cases and 2,865 controls that passed copy-number sample quality control.
Association testing was performed using a logistic regression model, where copy number was used as a predictor of early-onset MI. We included two principal components that estimated fine-scale population stratification as covariates in the model. Analyses were conducted using PLINK software.
To detect rare CNVs, we used a Hidden Markov Model, as implemented in the Birdseye package, and focused on rare (less than ~1% sample frequency) and large (greater than 100kb) autosomal deletions and duplications. Using methods recently described14, we evaluated case/control differences in rare CNVs across three parameters: genome-wide CNV rate, number of genes intersected by CNVs, and the total kilobase extent (see Supplementary Methods).
Statistical power
Given our inability to identify CNVs associated with MI, we estimated our statistical power for such discovery. For common CNVs, we had 78% power to detect a CNV of 25% frequency and effect size of 1.20 at an alpha of 0.001 in 3,000 cases and 3,000 controls.
For rare CNVs, we approximated by simulation the statistical power to detect a CNV with a population frequency for the deletion of 1/8000 (i.e., so it would be observed in 1/4,000 live births). We set the relative risk to 20.0 (i.e. the effect size seen for several rare variants associated with schizophrenia14) and the population disease prevalence to 1/100. We simulated 10,000 datasets for 2,920 cases and 3,035 controls under this model. Using Fisher’s exact test to account for small cell sizes, for a type I error rate of 0.01 (1-sided test) we had 97 % power. The mean case frequency was ~0.5%, the mean control frequency was ~0.02%. For a similarly rare variant but with a relative risk of 10.0, the average case frequency was ~0.25% (control frequency still ~0.02%) and power was lower at 54%.
These simulations suggest that we had good power to detect loci with large effects, although this assumes perfect sensitivity and specificity for detection. For very large deletions, at least, we expect sensitivity to detect such CNVs would be high. However, we may have missed additional loci with CNVs that are less penetrant, rarer, or smaller.
Supplementary Material
ACKNOWLEDGEMENTS
REGICOR. The REGICOR study was partially funded by the Ministerio de Sanidad y Consumo, Instituto de Salud Carlos III, the CIBER Epidemiología y Salud Pública, the FIS, and AGAUR Generalitat de Catalunya.
Massachusetts General Hospital. The MIGen study was funded by the U.S. National Institutes of Health (NIH) and National Heart, Lung, and Blood Institute’s STAMPEED genomics research program through a grant to D.A. S.K. is supported by a Doris Duke Charitable Foundation Clinical Scientist Development Award, a charitable gift from the Fannie E. Rippel Foundation, the Donovan Family Foundation, a career development award from the NIH, and institutional support from the Department of Medicine and Cardiovascular Research Center at Massachusetts General Hospital.
Broad Institute. Genotyping was partially funded by The Broad Institute Center for Genotyping and Analysis, which is supported by grant from the National Center for Research Resources.
FINRISK. V.S. was supported by the Sigrid Juselius Foundation.
WTCCC Study. The study was funded by the Wellcome Trust. Recruitment of cases for the WTCCC Study was carried out by the British Heart Foundation (BHF) Family Heart Study Research Group and supported by the BHF and the UK Medical Research Council. N.J.S. and S.G.B. hold Chairs funded by the BHF.
PennCATH/MedStar. Recruitment of the PennCATH cohort was supported by the Cardiovascular Institute of the University of Pennsylvania. Recruitment of the MedStar cohort was supported by a research grant from GlaxoSmithKline. Genotyping was performed at the Center for Applied Genomics at the Children’s Hospital of Philadelphia and supported by GlaxoSmithKline through an Alternate Drug Discovery Initiative research alliance award (to M.P.R. and D.J.R.) with the University of Pennsylvania School of Medicine. D.J.R. was supported by a Doris Duke Charitable Foundation Distinguished Clinical Scientist Award.
Verona Heart Study. The study was supported by a grant from the Italian Ministry of University and Research and grants from the Veneto Region and the Cariverona Foundation, Verona, Italy.
Mid-America Heart Institute. T.M. is supported by a career development grant from the NIH.
Irish Family Study. We thank the clinical staff members for their valuable contribution to the collection of families for this study. The research was supported by the Northern Ireland Research and Development Office, a Royal Victoria Hospital Research Fellowship, the Northern Ireland Chest, Heart and Stroke Association, and the Heart Trust Fund (Royal Victoria Hospital).
GerMIFS I and II. The German Study was supported by the Deutsche Forschungsgemeinschaft and the German Federal Ministry of Education and Researchin the context of the German National Genome Research Network.
Cardiogenics. Cardiogenics is an EU funded integrated project.
INTERHEART. S.A. holds the Michael G. DeGroote and Heart and Stroke Foundation of Ontario Chair in Population Health and the May Cohen Eli Lilly Endowed Chair in Women’s Health Research, McMaster University. We acknowledge the contribution Dr. S. Yusuf who initiated and together with the Steering Committee, supervised the conduct of the INTERHEART study. We thank members of the Project Office: S. Rangarajan (Study coordinator), and K. Hall (Laboratory manager) for their assistance in coordinating the genetics component of the INTERHEART project.
*. Myocardial Infarction Genetics Consortium
Manuscript preparation: Sekar Kathiresan (leader)1,2,3,4, Benjamin F. Voight2,3,5, Shaun Purcell2,3,6, Kiran Musunuru1,2,3,4, Diego Ardissino7, Pier M. Mannucci8, Sonia Anand9, James Engert10, Nilesh J Samani11, Heribert Schunkert12, Jeanette Erdmann12, Muredach P. Reilly13,14, Daniel J. Rader13,14, Thomas Morgan15, John Spertus16, Monika Stoll17, Domenico Girelli18, Pascal P McKeown19, Chris C Patterson19, David Siscovick20, Christopher J. O’Donnell1,4,21, Roberto Elosua22, Leena Peltonen3,23,24, Veikko Salomaa25, Stephen M. Schwartz20,26, Olle Melander27, David Altshuler2,3,4,5,28
Stage 1 discovery GWA studies:
Italian Atherosclerosis, Thrombosis, and Vascular Biology Study. Diego Ardissino, Pier Angelica Merlini29, Carlo Berzuni30, Luisa Bernardinelli30,31, Flora Peyvandi8, Marta Spreafico8, Marco Tubaro32, Patrizia Celli33, Maurizio Ferrario34, Raffaela Fetiveau34, Nicola Marziliano34, Giorgio Casari35, Michele Galli36, Flavio Ribichini37, Marco Rossi38, Francesco Bernardi39, Pietro Zonzin40, Alberto Piazza41, Rosanna Asselta42, Stefano Duga42, Pier M. Mannucci
Heart Attack Risk in Puget Sound. Stephen M. Schwartz, David S. Siscovick, Jean Yee20,26, Yechiel Friedlander43
Registre Gironi del COR. Roberto Elosua, Jaume Marrugat22, Gavin Lucas22, Isaac Subirana22, Joan Sala44, Rafael Ramos45
Massachusetts General Hospital Premature Coronary Artery Disease Study. Christopher J. O’Donnell, Sekar Kathiresan, Calum A. MacRae1,4
FINRISK. Veikko Salomaa, Aki S. Havulinna25, Leena Peltonen
Malmo Diet and Cancer Study. Olle Melander, Goran Berglund46
Stage 1 data analysis. Benjamin F. Voight (leader), Sekar Kathiresan, Joel N. Hirschhorn3,28, Kiran Musunuru, Mark Daly2,3,4, Shaun Purcell
Stage 1 phenotype data assembly. Stephen M. Schwartz (leader), Jean Yee, Sekar Kathiresan, Gavin Lucas, Isaac Subirana, Roberto Elosua
Stage 1 steering committee. Sekar Kathiresan, Diego Ardissino, Pier M. Mannucci, David Siscovick, Christopher J. O’Donnell, Roberto Elosua, Nilesh J. Samani, Leena Peltonen, Veikko Salomaa, Stephen M. Schwartz, David Altshuler (leader)
Stage 1 genome-wide genotyping. Aarti Surti3, Candace Guiducci3, Lauren Gianniny3, Daniel Mirel3, Melissa Parkin3, Noel Burtt3, Stacey Gabriel3 (leader)
Stage 2 replication studies:
Wellcome Trust Case Control Consortium. Nilesh J. Samani, John R. Thompson47, Peter S. Braund11, Benjamin J. Wright47, Anthony Balmforth48, Stephen G. Ball48, Alistair S. Hall48, and WTCCC* (*Please see Supplementary Materials for full listing of participants)
German MI Family Study I. Heribert Schunkert, Jeanette Erdmann, Patrick Linsel-Nitschke12, Wolfgang Lieb12, Andreas Ziegler49, Inke König49, Christian Hengstenberg50, Marcus Fischer50, Klaus Stark49, Anika Grosshennig12,49, Michael Preuss12,49, H.-Erich Wichmann51,52, Stefan Schreiber53
Cardiogenics. Heribert Schunkert, Nilesh J. Samani, Jeanette Erdmann, Willem Ouwehand54, Christian Hengstenberg, Panos Deloukas23, Michael Scholz55, Francois Cambien56
PennCATH/MedSTAR. Muredach Reilly, Mingyao Li57, Zhen Chen57, Robert Wilensky13,14, William Matthai14, Atif Qasim14, Hakon H. Hakonarson58, Joe Devaney59, Mary-Susan Burnett59, Augusto D. Pichard59, Kenneth M. Kent59, Lowell Satler59, Ron Waksman59, Steve Epstein59, Daniel Rader
Stage 3 replication studies:
Acute Myocardial Infarction Gene Study/Dortmund Health Study. Thomas Scheffold60, Klaus Berger61, Monika Stoll, Andreas Huge17
Verona Heart Study. Domenico Girelli, Nicola Martinelli18, Oliviero Olivieri18, Roberto Corrocher18
Mid-America Heart Institute. Thomas Morgan, John Spertus
Irish Family Study. Pascal P McKeown, Chris C Patterson
German MI Family Study II. Heribert Schunkert (leader), Jeanette Erdmann, Patrick Linsel-Nitschke, Wolfgang Lieb, Christian Hengstenberg, Marcus Fischer, Klaus Stark, Anika Grosshennig, Michael Preuss, H.-Erich Wichmann, Stefan Schreiber
INTERHEART. James C. Engert, Ron Do62, Changchun Xie9, Sonia Anand
Affiliations
Cardiovascular Research Center and Cardiology Division, Massachusetts General Hospital, Boston, Massachusetts 02114, USA
Center for Human Genetic Research, Massachusetts General Hospital, Boston, Massachusetts 02114, USA
Program in Medical and Population Genetics, Broad Institute of Harvard and Massachusetts Institute of Technology, Cambridge, Massachusetts 02142, USA
Department of Medicine, Harvard Medical School, Boston, Massachusetts 02115, USA
Department of Molecular Biology, Massachusetts General Hospital, Boston, Massachusetts, 02114, USA
Stanley Center for Psychiatric Research, Broad Institute of MIT and Harvard, Cambridge, Massachusetts 02142, USA
Division of Cardiology, Azienda Ospedaliero-Universitaria di Parma, Parma, Italy
Department of Internal Medicine and Medical Specialities, Fondazione Istituto di Ricovero e Cura a Carattere Scientifico, Ospedale Maggiore, Mangiagalli e Regina Elena, University of Milan, Milan, Italy
Population Health Research Institute, Hamilton Health Sciences and Departments of Medicine and Clinical Epidemiology and Biostatistics, McMaster University, Hamilton, Ontario, Canada.
Departments of Medicine and Human Genetics, McGill University, Montréal, Québec, Canada
Department of Cardiovascular Sciences, University of Leicester, Glenfield Hospital, LE3 9QP, UK
Medizinische Klinik II, Universität zu Lübeck, Lübeck, Germany
The Institute for Translational Medicine and Therapeutics, School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA
The Cardiovascular Institute, University of Pennsylvania, Philadelphia, Pennsylvania, USA
Department of Pediatrics, Vanderbilt University School of Medicine, Nashville, Tennessee, USA
Mid-America Heart Institute and University of Missouri-Kansas City, Kansas City, Missouri, USA
Leibniz-Institute for Arteriosclerosis Research, University Münster, Münster, Germany
Department of Clinical and Experimental Medicine, University of Verona, Verona, Italy
Centre for Public Health, Queen’s University Belfast, Institute of Clinical Science, Belfast, BT12 6BJ, Northern Ireland, UK
Cardiovascular Health Research Unit, Departments of Medicine and Epidemiology, University of Washington, Seattle, Washington, USA
Framingham Heart Study and National, Heart, Lung, and Blood Institute, Framingham, Massachusetts 01702, USA
Cardiovascular Epidemiology and Genetics, Institut Municipal D’investigacio Medica, and CIBER Epidemiología y Salud Pública, Barcelona, Spain.
Wellcome Trust Sanger Institute, Cambridge CB10 1SA, UK
Institute for Molecular Medicine, University of Helsinki, Helsinki 00029, Finland
Chronic Disease Epidemiology Unit, Department of Health Promotion and Chronic Disease Prevention, National Public Health Institute, Helsinki 00300, Finland
Department of Epidemiology, University of Washington, Seattle, Washington, USA
Department of Clinical Sciences, Hypertension and Cardiovascular Diseases, University Hospital Malmö, Lund University, Malmö 20502, Sweden
Department of Genetics, Harvard Medical School, Boston, Massachusetts 02115,USA
Division of Cardiology, Azienda Ospedaliera Niguarda Ca’ Granda, Milan, Italy
Biostatistics Unit, Medical Research Council, Cambridge, United Kingdom and Statistical Laboratory, Centre for Mathematical Sciences, Wilberforce Road, Cambridge, United Kingdom
Department of Applied Health Sciences, University of Pavia, Pavia, Italy
Division of Cardiology, Ospedale San Filippo Neri, Rome, Italy
Division of Cardiology, Ospedale San Camillo, Rome, Italy
Fondazione Istituto di Ricovero e Cura a Carattere Scientifico, Policlinico San Matteo, Pavia, Italy
Vita-Salute San Raffaele University and San Raffaele Scientific Institute, Milan, Italy
Division of Cardiology, Ospedale di Livorno, Livorno, Italy
Division of Cardiology, Ospedale Borgo Trento, University of Verona, Verona, Italy
Division of Cardiology, Istituto di Ricovero e Cura a Carattere Scientifico, Istituto Clinico Humanitas, Milan, Italy
Department of Biochemistry and Molecular Biology, University of Ferrara, Ferrara, Italy
Division of Cardiology, Ospedale di Rovigo, Rovigo, Italy
Department of Genetics, Biology and Biochemistry, University of Turin, Turin, Italy
Department of Biology and Genetics for Medical Sciences, University of Milan, 20133 Milan, Italy
Unit of Epidemiology, Hebrew University-Hadassah School of Public Health, Jerusalem, Israel
Servei de Cardiologia i Unitat Coronària, Hospital de Girona Josep Trueta and Institut de Investigació Biomedica de Girona, Spain
Unitat de Recerca i Unitat Docent de Medicina de Familia de Girona, IDIAP Jordi Gol, Institut Català de la Salut, Spain.
Department of Clinical Sciencs, Internal Medicine, University Hospital Malmö, Lund University, Malmö 20502, Sweden.
Department of Health Sciences, University of Leicester, Leicester, LE1 7RH, UK.
LIGHT and LIMM Research Institutes, Faculty of Medicine and Health, University of Leeds, Leeds, LS1 3EX, UK.
Institut für Medizinische Biometrie und Statistik, Universität zu Lübeck, Lübeck, Germany
Klinik und Poliklinik für Innere Medizin II, Universität Regensburg, Regensburg, Germany
Institute of Epidemiology, Helmholtz Zentrum München - German Research Center for Environmental Health, Neuherberg, Germany
Institute of Medical Information Science, Biometry and Epidemiology, LMU Munich, Germany
Institut für Klinische Molekularbiologie, Christian-Albrechts Universität, Kiel, Germany
Department of Haematology, University of Cambridge, Long Road, Cambridge, CB2 2PT, UK
Trium Analysis Online GmbH, Munich, Germany
INSERM UMR-S 525, UPMC Univ. Paris 06, Paris, France
Biostatistics and Epidemiology, University of Pennsylvania, Philadelphia, Pennsylvania, USA
The Center for Applied Genomics, Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania, USA
Cardiovascular Research Institute, MedStar Research Institute, Washington Hospital Center, Washington, DC 20010, USA.
Institute for Heart and Circulation Research of the University of Witten/Herdecke, Dortmund, Germany
Institute of Epidemiology and Social Medicine, University of Muenster, Germany
Department of Human Genetics, McGill University, Montréal, Québec, Canada
Footnotes
DISCLOSURES
The collection of clinical and sociodemographic data in the Dortmund Health Study was supported by the German Migraine- & Headache Society (DMKG) and by unrestricted grants of equal share from Astra Zeneca, Berlin Chemie, Boots Healthcare, Glaxo-Smith-Kline, McNeil Pharma (former Woelm Pharma), MSD Sharp & Dohme and Pfizer to the University of Muenster.
References
- 1.McPherson R, et al. A common allele on chromosome 9 associated with coronary heart disease. Science. 2007;316:1488–91. doi: 10.1126/science.1142447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Helgadottir A, et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science. 2007;316:1491–3. doi: 10.1126/science.1142842. [DOI] [PubMed] [Google Scholar]
- 3.Samani NJ, et al. Genomewide association analysis of coronary artery disease. N Engl J Med. 2007;357:443–53. doi: 10.1056/NEJMoa072366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nora JJ, Lortscher RH, Spangler RD, Nora AH, Kimberling WJ. Genetic--epidemiologic study of early-onset ischemic heart disease. Circulation. 1980;61:503–8. doi: 10.1161/01.cir.61.3.503. [DOI] [PubMed] [Google Scholar]
- 5.Rosamond W, et al. Heart disease and stroke statistics--2008 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2008;117:e25–146. doi: 10.1161/CIRCULATIONAHA.107.187998. [DOI] [PubMed] [Google Scholar]
- 6.Rissanen AM. Familial occurrence of coronary heart disease: effect of age at diagnosis. Am J Cardiol. 1979;44:60–6. doi: 10.1016/0002-9149(79)90251-0. [DOI] [PubMed] [Google Scholar]
- 7.Willer CJ, et al. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat Genet. 2008;40:161–9. doi: 10.1038/ng.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sebat J, et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–8. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- 9.Iafrate AJ, et al. Detection of large-scale variation in the human genome. Nat Genet. 2004;36:949–51. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 10.McCarroll SA, et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn’s disease. Nat Genet. 2008 doi: 10.1038/ng.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Willer CJ, Speliotes EK, Loos RJF, Li S, Lindgren CM, et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet. doi: 10.1038/ng.287. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weiss LA, et al. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med. 2008;358:667–75. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- 13.Walsh T, et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008;320:539–43. doi: 10.1126/science.1155174. [DOI] [PubMed] [Google Scholar]
- 14.Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–41. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu B, et al. Strong association of de novo copy number mutations with sporadic schizophrenia. Nat Genet. 2008;40:880–5. doi: 10.1038/ng.162. [DOI] [PubMed] [Google Scholar]
- 16.Stefansson H, et al. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–6. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCarroll SA, et al. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat Genet. 2008;40:1166–74. doi: 10.1038/ng.238. [DOI] [PubMed] [Google Scholar]
- 18.Korn JM, et al. Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat Genet. 2008;40:1253–60. doi: 10.1038/ng.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cohen JC, Boerwinkle E, Mosley TH, Jr., Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354:1264–72. doi: 10.1056/NEJMoa054013. [DOI] [PubMed] [Google Scholar]
- 20.Pe’er I, Yelensky R, Altshuler D, Daly MJ. Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genet Epidemiol. 2008;32:381–5. doi: 10.1002/gepi.20303. [DOI] [PubMed] [Google Scholar]
- 21.Kathiresan S, et al. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat Genet. 2008;40:189–97. doi: 10.1038/ng.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Allen PB, Greenfield AT, Svenningsson P, Haspeslagh DC, Greengard P. Phactrs 1-4: A family of protein phosphatase 1 and actin regulatory proteins. Proc Natl Acad Sci U S A. 2004;101:7187–92. doi: 10.1073/pnas.0401673101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cavdar Koc E, Burkhart W, Blackburn K, Moseley A, Spremulli LL. The small subunit of the mammalian mitochondrial ribosome. Identification of the full complement of ribosomal proteins present. J Biol Chem. 2001;276:19363–74. doi: 10.1074/jbc.M100727200. [DOI] [PubMed] [Google Scholar]
- 24.Kwon HM, et al. Cloning of the cDNa for a Na+/myo-inositol cotransporter, a hypertonicity stress protein. J Biol Chem. 1992;267:6297–301. [PubMed] [Google Scholar]
- 25.Abbott GW, et al. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell. 1999;97:175–87. doi: 10.1016/s0092-8674(00)80728-x. [DOI] [PubMed] [Google Scholar]
- 26.Schunkert H, et al. Repeated replication and a prospective meta-analysis of the association between chromosome 9p21.3 and coronary artery disease. Circulation. 2008;117:1675–84. doi: 10.1161/CIRCULATIONAHA.107.730614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hannon GJ, Beach D. p15INK4B is a potential effector of TGF-beta-induced cell cycle arrest. Nature. 1994;371:257–61. doi: 10.1038/371257a0. [DOI] [PubMed] [Google Scholar]
- 28.Ridker PM, Rifai N, Cook NR, Bradwin G, Buring JE. Non-HDL cholesterol, apolipoproteins A-I and B100, standard lipid measures, lipid ratios, and CRP as risk factors for cardiovascular disease in women. JAMA. 2005;294:326–33. doi: 10.1001/jama.294.3.326. [DOI] [PubMed] [Google Scholar]
- 29.Martinelli N, et al. FADS genotypes and desaturase activity estimated by the ratio of arachidonic acid to linoleic acid are associated with inflammation and coronary artery disease. Am J Clin Nutr. 2008;88:941–9. doi: 10.1093/ajcn/88.4.941. [DOI] [PubMed] [Google Scholar]
- 30.Morgan TM, Krumholz HM, Lifton RP, Spertus JA. Nonvalidation of reported genetic risk factors for acute coronary syndrome in a large-scale replication study. JAMA. 2007;297:1551–61. doi: 10.1001/jama.297.14.1551. [DOI] [PubMed] [Google Scholar]
- 31.Meng W, et al. Genetic variants of complement factor H gene are not associated with premature coronary heart disease: a family-based study in the Irish population. BMC Med Genet. 2007;8:62. doi: 10.1186/1471-2350-8-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Serre D, et al. Correction of population stratification in large multi-ethnic association studies. PLoS ONE. 2008;3:e1382. doi: 10.1371/journal.pone.0001382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zeggini E, et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet. 2008;40:638–45. doi: 10.1038/ng.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analysis. Am J Hum Genet. 2007;81:3. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schadt EE, et al. Mapping the genetic architecture of gene expression in human liver. PLoS Biol. 2008;6:e107. doi: 10.1371/journal.pbio.0060107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.