

Abstract
Recently, a requirement for β-arrestin–mediated endocytosis in the activation of extracellular signal–regulated kinases 1 and 2 (ERK1/2) by several G protein–coupled receptors (GPCRs) has been proposed. However, the importance of this requirement for function of ERK1/2 is unknown. We report that agonists of Gαq-coupled proteinase–activated receptor 2 (PAR2) stimulate formation of a multiprotein signaling complex, as detected by gel filtration, immunoprecipitation and immunofluorescence. The complex, which contains internalized receptor, β-arrestin, raf-1, and activated ERK, is required for ERK1/2 activation. However, ERK1/2 activity is retained in the cytosol and neither translocates to the nucleus nor causes proliferation. In contrast, a mutant PAR2 (PAR2δST363/6A), which is unable to interact with β-arrestin and, thus, does not desensitize or internalize, activates ERK1/2 by a distinct pathway, and fails to promote both complex formation and cytosolic retention of the activated ERK1/2. Whereas wild-type PAR2 activates ERK1/2 by a PKC-dependent and probably a ras-independent pathway, PAR2(δST363/6A) appears to activate ERK1/2 by a ras-dependent pathway, resulting in increased cell proliferation. Thus, formation of a signaling complex comprising PAR2, β-arrestin, raf-1, and activated ERK1/2 might ensure appropriate subcellular localization of PAR2-mediated ERK activity, and thereby determine the mitogenic potential of receptor agonists.
Keywords: subcellular localization, MAP kinase, cytosol, receptor trafficking
Introduction
Agonists of certain G protein–coupled receptors (GPCRs) activate extracellular signal–regulated kinases 1 and 2 (ERK1/2), but do not induce their translocation into the nucleus (Pouyssegur and Böhm 1992; Alblas et al. 1996; Post et al. 1996). Although the mitogenic effects of nuclear ERK1/2 are the most often studied, a number of important substrates of ERK1/2 are cytoplasmic. Some examples are insulin receptor substrate-1, where phosphorylation by ERK1/2 leads to cellular insulin resistance (DeFea and Roth 1997), cytoskeletal proteins, and microtubule-associated proteins (MAPs; Sturgill and Ray 1986; Ray and Sturgill 1988), where phosphorylation is involved in cell migration, morphological alterations (Goedert et al. 1996; Klemke et al. 1997), and phospholipase A2 (Bornfeldt et al. 1997), which mediates many agonist-induced inflammatory responses. In addition, some of the transcription factors that are activated by ERK1/2 are cytoplasmic, and translocate to the nucleus after phosphorylation (Chen et al. 1992). A number of stimuli converge on the same ERK cascade to elicit specific biological responses. If a given agonist is intended to induce ERK-dependent phosphorylation of cytosolic substrates, but not induce proliferation, how does the cell ensure that ERK activation results in the correct response? An answer may lie in protein scaffolding complexes, such as those observed for the β2-adrenergic receptor (β2-AR). In this instance, a signaling complex in clathrin-coated endocytic vesicles comprising activated receptor, the clathrin adaptor protein β-arrestin, and Src, is required for ERK activation (Daaka et al. 1998; Luttrell et al. 1999). Similar requirements have been found for other GPCRs (Ignatova et al. 1999; Vögler et al. 1999). However, none of these studies have addressed the question of why the β-arrestin–bound, internalized receptor would be required for activation of ERK1/2 when other signals transduced by the receptor occur while it is still at the cell surface.
The purpose of the present investigation was to define the role of β-arrestin–containing scaffolding complexes in the activation and subcellular localization of ERK1/2. We examined the role of these complexes in signal transduction by proteinase-activated receptor-2 (PAR2), a member of a new family of GPCRs for serine proteases (Déry et al. 1998). Pancreatic trypsin and mast cell tryptase cleave PAR2 within its extracellular NH2 terminus (SKGR ↓ SLIGRL/KV, ↓ cleavage site for rat/human PAR2), exposing a tethered ligand domain (SLIGRL/KV) that binds to and activates the cleaved receptor. PAR2 is highly expressed in the gastrointestinal tract and pancreas. Pancreatic trypsin activates PAR2 in enterocytes and in epithelial cells of the pancreatic duct to regulate prostanoid secretion and the activity of ion channels (Kong et al. 1997; Nguyen et al. 1999). Tryptase, a major protease of human mast cells, activates PAR2 in enteric myocytes and neurons, and PAR2 may mediate some of the mitogenic and proinflammatory effects of tryptase (Corvera et al. 1997, Corvera et al. 1999). In view of the potential importance of PAR2 in normal regulation, inflammation and mitogenesis (Déry et al. 1998), it is important to understand the mechanisms of PAR2 signal transduction. PAR2 couples Gαq/11 and phospholipase Cβ, leading to hydrolysis of phosphatidylinositol bisphosphate, Ca2+ mobilization, and activation of protein kinase C (PKC) and ERK1/2 (Déry et al. 1998). Agonist-induced desensitization of PAR2 involves receptor phosphorylation and endocytosis by a β-arrestin and clathrin-dependent mechanism (Böhm et al. 1996; Déry et al. 1999).
We investigated the role of β-arrestins as molecular scaffolds that organize and recruit components of the mitogen-activated protein (MAP) kinase cascade, and thereby ensure the proper localization and specificity of activated ERK1/2. Our aims were as follows: to determine if β-arrestin–dependent receptor endocytosis is required for PAR2-mediated ERK activation; identify components of this pathway that interact with β-arrestins; investigate whether a desensitization and internalization-resistant PAR2 mutant, which does not interact with β-arrestins, activates ERK1/2 by alternative pathways; and examine whether the formation of the complex affects the localization and specificity of ERK1/2.
Materials and Methods
Materials
Unless otherwise specified, all reagents were from Sigma Chemical Co. Trypsin (TPCK-treated) was from Worthington Biochemical, Inc. Peptides corresponding to the tethered ligand domains of rat and human PAR2 (activating peptides, AP: rat SLIGRL-NH2; human SLIGKV-NH2) were synthesized on solid phase and purified by high pressure liquid chromatography. SLIGKV-NH2 was used as an agonist of transfected cells expressing human PAR2, and SLIGRL-NH2 was used to stimulate rat enterocytes. Fura-2/AM and propidium iodide were from Molecular Probes, Inc. An expression vector for enhanced green fluorescent protein (pEGFP-N1) was from CLONTECH Laboratories. A Geneditor kit was from Promega Corp. Expand polymerase was from Boehringer Mannheim. Phosphocellulose columns and enhanced chemiluminescence detection kits were from Pierce Chemical Co. PVDF membranes were from New England Nuclear. 4α-phorbol 12,13-dicanoate (PDB) was from Calbiochem-Novabiochem Corp. Protein A–agarose and Lipofectin were from GIBCO BRL. β-Arrestin-1 cDNA was a gift from Dr. R.J. Lefkowitz (Duke University, Durham, NC). pCEP-ERK2 was from Dr. M. Cobb (University of Texas, Dallas, TX). N17ras was from Dr. A. Weiss (University of California, San Francisco, San Francisco, CA). Kirsten murine sarcoma virus–transformed rat kidney epithelial cells (KNRK) were from the American Type Tissue Culture Collection (ATCC CRL 1569). A rat enterocyte cell line hBRIE380, which highly expresses PAR2 (26), was from Dr. G. Aponte (University of California, Berkeley, Berkeley, CA). HEK293 cells were from Dr. R. Roth (Stanford University, Stanford, CA).
Antibodies
The following antibodies were used: phosphorylated ERK1/2 (pERK1/2), total ERK1/2 conjugated to agarose beads, c-src, Shc, phosphotyrosine (PY-99), and G-actin (Santa Cruz Biotechnology); total ERK1/2 (Zymed Labs, Inc.); β-arrestin-1, raf-1, PYK2, and histone (Transduction Labs); β-arrestin-1/2 (a gift from Dr. R.J. Lefkowitz); enhanced green fluorescent protein (EGFP; McConalogue et al. 1998) (a gift from Dr. J.H. Walsh, University of California, Los Angeles, CA); Flag M1 (International Biotechnologies Inc.); HA.11 to the hemagglutinin 12CA5 epitope (YPYDVPDYA) Berkeley Antibody Co.; HA.11 conjugated to HRP, Boehringer. Secondary antibodies coupled to FITC and Texas red were from Jackson ImmunoResearch Laboratories, Inc. A secondary antibody conjugated to (R)-phycoerythrin was from Caltag Laboratories. A secondary antibody conjugated to HRP was from Pierce Chemical Co.
A rabbit polyclonal antibody (PAR2N) raised to an NH2-terminal fragment of human PAR2 (↓37SLIGKVDGTSHVTGKG53V, ↓ trypsin cleavage site) was from Dr. P. Andrade-Gordon (Johnson and Johnson, PRI). This antibody detected a single protein of 80 kD on Western blots of hBRIE cells, and proteins of 49 and 78 kD on blots of KNRK cells expressing human PAR2 with a 12CA5 epitope (Böhm et al. 1996). These proteins were not detected in untransfected KNRK cells, which express PAR2 only at very low levels. The signals were abolished by preincubation of the antibody with the receptor fragment used for immunization. The 78-kD protein represents glycosylated PAR2 as it can be reduced to 49 kD (corresponding to the mass of PAR2) by with endoglycosidase. Further, both proteins cross-reacted with an antibody to the 12CA5 epitope. A rabbit polyclonal antiserum (B5) raised to a peptide corresponding to rat PAR2 (30GPNSKGR↓SLIGRLDT46P-YGGC) was from Dr. M. Hollenberg (University of Calgary, Alberta, Canada; Kong et al. 1997).
Plasmid Construction and Generation of Cell Lines
KNRK cells were stably transfected with human PAR2 alone, or with PAR2 plus rat β-arrestin-1 tagged with COOH-terminal green fluorescent protein (GFP, designated ARR-GFP), or a fragment of β-arrestin319-418 tagged with GFP (ARR319-418-GFP), as described (Déry et al. 1999). This fragment corresponds to the clathrin-binding domain of β-arrestins, and is a dominant negative mutant for β-arrestin–mediated endocytosis. The human PAR2 construct comprised NH2-terminal Flag proximal to the trypsin cleavage site and COOH-terminal 12CA5 epitopes (Böhm et al. 1996). For some experiments hBRIE cells were stably transfected with ARR-GFP or with ARR319-418-GFP using Lipofectin (Déry et al. 1999). The point mutant PAR2(δST363/6A) was created using the Geneditor kit according to manufacturer's instructions. The mutagenic oligonucleotide (OPERON Technologies) contained a Dde1 site, which allowed identification of mutant PAR2 by restriction digestion. DNA was sequenced to confirm the mutation. KNRK cells were stably transfected with PAR2(δST363/6A) alone or cotransfected with either ARR-GFP or ARR319-418-GFP using Lipofectin, and were selected and maintained as described for KNRK-PAR2 cells (Déry et al. 1999). HEK293 cells were transfected with human PAR2 alone, PAR2 plus ARR-GFP or ARR319-418-GFP, or with PAR2δST363/6A using Lipofectin. Stable cell lines were selected and maintained as described (Déry et al. 1999). Rat-ERK2 was amplified from pCEP-ERK2 by PCR and ligated into pEGFP-N1 at Bgl2 and Age1 sites, to generate ERK2-GFP. DNA was sequenced to confirm that the fusion junction was in-frame. To visualize translocation of ERKs, KNRK-PAR2 and KNRK-PAR2(δST363/6A) cells were transiently transfected with ERK2-EGFP using calcium phosphate, as described (DeFea and Roth 1997). To examine the role of ras in ERK activation, KNRK-PAR2 and KNRK-PAR2(δST363/6A) cells were stably transfected with dominant negative pcDNA3.1-N17ras using Lipofectin.
Two to three individual clones of each cell line were screened for expression of PAR2 or β-arrestins by measuring Ca2+ mobilization in response to PAR2 agonists, by flow cytometry and Western blotting. Selected clones expressed PAR2 or β-arrestins at similar levels and, thus, single clones were used in the remainder of the studies. Untransfected KNRK cells did not express significant levels of PAR2, as demonstrated by lack of Ca2+ mobilization in response to PAR2 agonists and lack of immunoreactivity to PAR2 on Western blots. Levels of endogenous β-arrestins were slightly higher in KNRK and 293 cells than in hBRIE cells as detected by Western blotting.
MAPK Assays
Confluent cells were maintained in minimal essential medium without serum overnight and incubated with 50 nM trypsin or 50 μM AP for 0–60 min at 37°C. Cells were incubated with 100 nM GF109203X, 20 nM LY379196, 10 μM genistein, and 20 μM tyrphostin-25 for 20 min or with 100 ng/ml pertussis toxin (PTX) for 24 h. Vehicle was used for controls. Cells were lysed in RIPA (PBS with 1% NP-40, 0.25% sodium deoxycholate, 400 nM okadaic acid, and 2 mM orthovanadate and protease inhibitors), and ERK1/2 were immunoprecipitated by incubating 300 μg total protein with ERK antibodies coupled to agarose beads. For measurement of ERK1/2 activity, beads were washed and incubated with 5 mM myelin basic protein (MBP) in the presence of 1 μCi γ[32P]ATP and 10 μM cold ATP in 50 mM Tris-HCl, pH 7.6, 6 mM MgCl2, and 2 mM MnCl2 for 30 min at room temperature. Reactions were stopped by centrifugation and an addition of an equal volume of Laemmli sample buffer to the supernatant or by binding MBP to a phosphocellulose column. Samples were analyzed by 20% SDS-PAGE and autoradiography. Phosphorylation was assessed by counting excised MBP bands or phosphocellulose-bound MBP. All counts were normalized to the MBP band density and immunoprecipitated ERK1/2. MBP was identified by Coomassie staining of gels. To detect total immunoprecipitation of ERK1/2, the immunoprecipitates were boiled, analyzed by 12% SDS-PAGE, transferred to PVDF membranes, and incubated with antibody to total ERK1/2 (1:1,000, overnight, 4°C), followed by goat anti–rabbit IgG conjugated to HRP (1:20,000 for 1 h at room temperature). Proteins were visualized by enhanced chemiluminescence. Autoradiograms and Coomassie-stained gels were photographed using a digital camera, and images were analyzed using Adobe Photoshop 4.0. Band density was determined by histogram analysis (mean density × number pixels). The total counts per minute present in each excised band was normalized to both the density of the MBP band and the ERK1/2 bands representing the amount of ERK present in each reaction. Activation of ERK1/2 was also assessed by Western blotting using antibodies to phospho-ERKs. 10 μg of total protein was removed before immunoprecipitation, analyzed by 12% SDS-PAGE, and transferred to PVDF membranes. Membranes were incubated with pERK antibody (1:400, overnight, 4°C), followed by goat anti–mouse IgG conjugated to HRP (1:30,000, for 1 h at room temperature). Blots were stripped in 2% SDS/1 mM β-mercaptoethanol in 50 mM Tris-HCl, pH 6.8, 150 mM NaCl for 60 min, washed, and reprobed with antibody to total ERK1/2 to ensure equal levels of ERK1 and 2 were present at each time point. Results were normalized as described for the total amount of ERK 1 and 2 in each reaction. Phosphorylation was assessed by histogram analysis of both ERK1 and ERK2 band densities (mean density × number pixels) and fold phosphorylation over basal was calculated. Fold phosphorylation of ERK1 and ERK2 was not substantially different; thus, ERK1 and 2 band densities were added together and were represented as ERK1/2 phosphorylation.
Subcellular Fractionation
For subcellular localization of pERK, cells were treated as described, ruptured in hypotonic buffer (HPB: 50 mM Hepes, pH 7.6, 1 mM MgCl2, 2 mM orthovanadate, 500 nM okadaic acid, and protease inhibitors) with 25 strokes in a glass Dounce homogenizer, and centrifuged at 800 g for 10 min. Supernatants were cleared at 100,000 g, and the cytosolic protein was precipitated with 10 vol of ice-cold acetone. Cytosolic protein pellets and plasma membrane pellets were resuspended in HPB + 0.1% NP-40. Nuclei-containing pellets from the low-speed spin were washed in HPB, resuspended in HPB + 0.1% NP-40 and pelleted at 10,000 g. Nuclear supernatants were precipitated with 10% ice-cold acetone and saved for analysis. Nuclei were resuspended in Laemmli buffer and sonicated for 30 s. pERK was determined in cytosolic and nuclear pellets by Western blotting. The identity of nuclear and cytosolic fractions was confirmed by Western blotting with antibodies to histone and G-actin, respectively.
Immunoprecipitation
Cells were maintained overnight in minimal essential medium without serum overnight, incubated with 50 nM trypsin or 50 μM AP for 0–30 min at 37°C, and lysed in RIPA. For KNRK-PAR2, KNRK-PAR2(δST363/6A), and KNRK-PAR2+ARR319-418-GFP cells, 500 μg total protein was incubated with either 1 μg of antibody to β-arrestin-1/2, 500 ng antibody to c-src, 1 μg antibody to PYK2, or 500 ng antibody to Shc. For hBRIE-ARR-GFP, KNRK-PAR2+ARR-GFP, KNRK-PAR2+ARR319-418-GFP, and KNRK-PAR2(δST363/6A)+ARR-GFP cells, 500 μg total protein was immunoprecipitated with 2 μg of antibody to GFP. Polyclonal antibodies were prebound to 40 μl of protein A–agarose for 2 h at 4°C. mAbs were incubated with equal concentrations of rabbit anti–mouse IgG before binding protein A. Beads were washed twice in TBS (50 mM Tris-HCl, pH 8.0, and 150 mM NaCl), and boiled in Laemmli sample buffer. Proteins were analyzed by 10% SDS-PAGE, followed by Western blotting with antibodies to raf-1 (1:300) or phosphotyrosine (1:500). Blots were stripped and reprobed with monoclonal β-arrestin-1 antibody (1:1,000) to ensure equal levels of β-arrestin-1 in all lanes or with PYK2 (1:1,000) or Shc (1:500) antibodies to confirm equal levels and identity of phosphoproteins.
Gel Filtration
Cells were lysed in RIPA as described. Lysates were loaded onto a column (3 × 150 cm, 400 ml bed volume) of S300 Sephacryl preequilibrated in PBS. Proteins were eluted in PBS at 1.1 ml/min, and fractions were collected at 7-min intervals. The column was calibrated with Dextran blue, 3 M KCl, 8.5 nm thyroglobulin, 5.2 nm catalase, 3.5 nm BSA, and 1.8 nm myoglobin. Absorbance of each fraction was measured at 290 nm. Proteins in the fractions from the included volume were precipitated with methanol/chloroform and analyzed by 10% SDS-PAGE, followed by Western blotting with antibodies to PAR2 (HA.11 or 2N), β-arrestin-1, raf-1, pERK, and total ERK1/2, as described. The band densities for each protein were divided by the sum of the band densities of all fractions (100%) in the included volume to determine the percentage of the total protein eluting in each fraction. Partition coefficients (σ), error function complements of σ, and Stoke's radii were calculated according to the method of Ackers (Chun et al., 1969). Fractions that were found to contain all four proteins were pooled and concentrated 30-fold by dialysis against solid sucrose at 4°C, followed by dialysis against PBS overnight at 4°C. Concentrated proteins were immunoprecipitated with antibodies to β-arrestin-1/2 (1 μg/ml, bound to protein A-agarose) or pERK1/2 (1 μg/ml bound to protein A–agarose).
Immunofluorescence and Confocal Microscopy
Cells grown on glass coverslips were maintained in minimal essential medium without serum overnight and incubated with 50 nM trypsin for 0–30 min at 37°C. Cells were fixed in 4% paraformaldehyde in 100 mM PBS, pH 7.4, for 20 min at 4°C, washed in PBS, and incubated in PBS containing 0.5% Tween and 1% BSA for 30 min. PAR2 was localized using a mouse monoclonal HA.11 antibody (1:5,000, overnight, 4°C; Déry et al. 1999). β-Arrestin-1 was detected using GFP (in KNRK-PAR2+ARR-GFP cells) or by immunofluorescence using an antibody to β-arrestin-1+2 (1:1,000, overnight, 4°C, KNRK-PAR2(δST363/6A) cells) (Déry et al. 1999). Raf-1 was localized by immunofluorescence using a mouse mAb (1:500, overnight, 4°C). Secondary antibodies coupled to FITC (for β-arrestin-1+2) or Texas red (for HA.11 and raf-1) were used (1:200, for 2 h at room temperature). Cells were observed using a Bio-Rad MRC 1000 laser scanning confocal microscope, a Zeiss Axiovert 100× microscope, and a Zeiss 100× Plan-Apochromat oil immersion objective (NA 1.4; Déry et al. 1999). For observations of trafficking in real time, KNRK-PAR2 and KNRK-PAR2(δST363/6A) cells transiently expressing ERK2-GFP were maintained at 37°C in a microincubator. Care was taken to minimize laser exposure by limiting the number of optical sections (2–3 per time point) and the laser intensity (<3%, NA 3–4 mm), to maintain cell viability (Déry et al. 1999). Parallel time points were fixed, stained with DAPI, and observed using a Zeiss Axioplan fluorescence microscope with a Zeiss 100× Plan-Apochromat oil immersion objective to confirm nuclear localization.
Flow Cytometry
Flow cytometry was used to quantify removal of PAR2 from the plasma membrane and to thereby determine the rate of endocytosis. Cells were incubated with 50 μM AP for 0–30 min at 37°C, and surface Flag immunoreactivity was quantified by flow cytometry (Déry et al. 1999). Like trypsin, AP induces PAR2 endocytosis (Böhm et al. 1996) but without removing the Flag epitope. Transfected cells were incubated with 10 μg/ml M1 antibody for 1 h at 4°C, washed, and incubated with 2 μg/ml phycoerythrin-conjugated goat anti–mouse IgG for 1 h at 4°C. hBRIE cells were incubated with 2 μg/ml B5 antibody for 1 h at 4°C, washed, and incubated with 2 μg/ml FITC-conjugated goat anti–rabbit IgG for 1 h at 4°C. Surface fluorescence was analyzed with a Facscan flow cytometer (Becton Dickinson Co.). Fluorophores were excited at 488 nm and emission was collected at 575 nm for phycoerythrin and 530 nm for FITC. Viability was assessed by exclusion of propidium iodide.
Measurement of [Ca2+]i
Cells were incubated in physiological salt solution (PSS [in mM]: 137 NaCl, 4.7 KCl, 0.56 MgCl2, 2 CaCl2, 1.0 Na2HPO4, 10 N-2-hydroxyethyl piperazine-N′-2-ethanesulphonic acid, 2.0 l-glutamine, and 5.5 d-glucose, pH 7.4) containing 0.1% BSA and 5 μM Fura-2/AM for 20 min at 37°C. They were washed and mounted in a microincubator containing 1 ml of PSS-BSA at 37°C on the stage of a Zeiss Axiovert 100 TV microscope. Agonists were directly added to the bath. Cells were observed with a Zeiss Fluar 40× objective (NA 1.30) and fluorescence was detected in individual cells using an ICCD video camera (Stanford Photonics) and a video microscopy acquisition program (Axon Instruments; Corvera et al. 1997, Corvera et al. 1999). Fluorescence was measured at 340 and 380 nm excitation and 510 nm emission. The ratio of the fluorescence at the two excitation wavelengths, which is proportional to the [Ca2+]i, was determined. For concentration-response experiments, cells were exposed to a single concentration of agonist. To examine homologous desensitization, cells were repeatedly challenged with agonists without an intervening wash (Böhm et al. 1996). To examine the role of PKC in desensitization, cells were incubated with 1 μM PDB for 20 min before agonist challenge (Böhm et al. 1996).
Proliferation Assays
KNRK-PAR2, KNRK-PAR2(δST363/6A), and hBRIE cells were seeded at 104 cells/well in 24-well plates, grown to 80% confluence, and maintained in minimal essential medium without serum overnight. Cells were incubated with 50 μM AP or 20% FCS for 24 h at 37°C. During the last 20 h, 1 μCi [3H]thymidine was added. Cells were washed, DNA was obtained by solubilization in 0.2 M NaOH, and incorporated [3H]thymidine was measured by scintillation counting. Cells were also counted in a hemacytometer. Results are expressed as the fold increase over untreated cells.
Statistical Analyses
Results are expressed as mean ± standard error. Differences between multiple groups were examined by ANOVA and Student-Newman-Keuls test, with P < 0.05 considered to be significant.
Results
Expression of Dominant Negative β-Arrestin Inhibits PAR2-mediated ERK Activity
We have previously shown that expression of β-arrestin319-418 prevents agonist-induced endocytosis of PAR2, and that overexpression of ARR-GFP has no effect on endocytosis or on the magnitude and duration of Ca2+ signaling (Déry et al. 1999). To examine the importance of β-arrestin–mediated receptor endocytosis for activation of ERK1/2, we coexpressed human PAR2 (NH2-terminal Flag, COOH-terminal HA.11 epitopes) with either β-arrestin-1 fused to green fluorescent protein (ARR-GFP) or with a dominant negative fragment of β-arrestin fused to GFP (ARR319-418-GFP) in KNRK cells (Déry et al. 1999). We assayed ERK activity using MBP as a substrate. In KNRK-PAR2+ARR-GFP cells, 50 nM trypsin stimulated a peak ERK activity of 5.76 ± 0.8-fold over basal at 5 min (Fig. 1 a, •). In contrast, in KNRK-PAR2+ARR319-418-GFP cells, ERK1/2 activation was significantly diminished to only 1.8 ± 0.3-fold over basal at 5 min, and trypsin only induced a significant elevation of 2.3 ± 0.3-fold after 30 min (P < 0.05; Fig. 1 a, ○). To ensure that trypsin-stimulated MBP activity was not an artifact of PAR2 overexpression in transfected cells, we repeated the experiment in hBRIE cells, an enterocyte cell line naturally expressing PAR2 (Kong et al. 1997). In hBRIE cells, 50 nM trypsin stimulated peak of 4.4 ± 1-fold over basal at 5 min (Fig. 1 a, □).
Figure 1.

PAR2-mediated activation of ERK1/2. (a) KNRK-PAR2+ARR-GFP cells (•) and KNRK-PAR2+ARR319-418-GFP cells (○) and hBRIE cells (□) were incubated with 50 nM trypsin for 0–60 min at 37°C, and ERK activity was measured using the MBP assay. (b–d) Western blots using antibodies to pERK1/2. (b) KNRK-PAR2+ARR-GFP cells (•), KNRK-PAR2 cells (⋄), and KNRK-PAR2+ARR319-418-GFP cells (○) were incubated with 50 nM trypsin. (c) hBRIE cells (□), hBRIE+ARR-GFP (⋄), and hBRIE+ARR319-418-GFP (♦) were incubated with 50 nM trypsin for 0–30 min at 37°C. (d) KNRK-PAR2+ARR-GFP cells (•) and KNRK-PAR2+ARR319-418-GFP cells (○) were incubated 50 μM AP for 0–30 min at 37°C. *P < 0.05 compared with cells expressing PAR2 alone or PAR2 plus ARR-GFP cells, n = 4.
Similar results were obtained using pERK antibodies to detect activated, phosphorylated ERK1/2. Thus, 50 nM trypsin maximally stimulated a 5.0 ± 0.3-fold increase in ERK1/2 phosphorylation in KNRK-PAR2+ARR-GFP cells (Fig. 1 b, •), but only a 1.2 ± 0.4-fold increase in KNRK-PAR2+ARR319-418-GFP cells (Fig. 1 b, ⋄). Similarly, 50 nM trypsin maximally stimulated a 5.1 ± 0.4-fold increase in ERK1/2 phosphorylation in KNRK-PAR2 cells (Fig. 1 b, ⋄). Comparable results were obtained in enterocytes, where 50 nM trypsin stimulated maximal ERK1/2 phosphorylation by 5.0 ± 0.3-fold in hBRIE cells (Fig. 1 c, □), 4.85 ± 0.45-fold in hBRIE+ARR-GFP cells (Fig. 1 c, ▪), but by only 1.5 ± 0.2-fold in hBRIE+ARR319-419-GFP cells (Fig. 1 c, ♦).
To ensure that the effect of trypsin on ERK1/2 activation was due to PAR2 stimulation and not to other effects of trypsin on the cell, we examined the ability of the AP corresponding to the PAR2-tethered ligand to stimulate ERK1/2. In KNRK-PAR2+ARR-GFP cells, 50 μM AP stimulated a 4.8 ± 0.5-fold increase in ERK phosphorylation that was not observed in KNRK-PAR2+ARR319-418-GFP cells (Fig. 1 d). Similar results were observed with hBRIE cells (not shown).
These results were confirmed in a human embryonic kidney fibroblast cell line, HEK293 cells, where trypsin stimulated a 5.0 ± 0.2 and a 5.3 ± 0.6-fold increase in ERK1/2 phosphorylation in cells expressing PAR2 alone or PAR2+ARR-GFP, respectively. Coexpression of ARR319-418-GFP with PAR2 inhibited trypsin-stimulated ERK1/2 activation in HEK 293 cells by 90 ± 10% (not shown). Phorbol ester–stimulated ERK1/2 phosphorylation was unaffected by expression of β-arrestin319-418, indicating that there is not a basic requirement for β-arrestin in all mechanisms of MAPK activation (not shown).
Together, these results indicate that PAR2 agonists activate ERK1/2 in transfected cell lines (KNRK and HEK) and in enterocytes (hBRIE) that naturally express this receptor. Expression of β-arrestin319-418, which we have previously shown to suppress agonist-induced endocytosis of PAR2 (Déry et al. 1999), inhibits PAR2-mediated ERK1/2 activation in both transfected cells and in hBRIE cells. However, overexpression of β-arrestin does not affect this response (Fig. 1, b–d).
An Internalization-resistant and Desensitization-defective PAR2 Mutant Activates ERK1/2
We have previously reported that PAR2-induced Ca2+ mobilization is rapidly attenuated and desensitized (Böhm et al. 1996). The mechanism of desensitization probably involves phosphorylation of the PAR2 intracellular C-tail by G protein receptor kinases and PKC, and association with β-arrestins, which uncouple the receptor from heterotrimeric G proteins and also serve as clathrin adaptors for endocytosis (Böhm et al. 1996; Déry et al. 1999). β-Arrestin319-418 corresponds to the clathrin binding domain of β-arrestins. Expression of this mutant prevents PAR2 internalization but does not affect desensitization, so that KNRK-PAR2+ARR319-418-GFP cells signal normally, and this signal is rapidly desensitized. Moreover, overexpression of ARR-GFP does not affect PAR2 internalization, desensitization, or Ca2+ mobilization (Déry et al. 1999).
To examine the requirement of receptor endocytosis and desensitization for activation of ERK1/2, we mutated several potential phosphorylation sites in the cytoplasmic tail of PAR2. A double point mutant, in which serine and threonine at codons 363 and 366 were changed to alanines PAR2(δST363/6A), showed markedly reduced receptor desensitization and internalization. Trypsin stimulated Ca2+ mobilization in both KNRK-PAR2 and KNRK-PAR2(δST363/6A) cells with a similar efficacy, although with a higher potency in KNRK-PAR2(δST363/6A) cells (Fig. 2 a). The duration of activation was measured as the time during which Ca2+ mobilization was >200% of the baseline. In KNRK-PAR2(δST363/6A) cells, 10 nM trypsin induced a Ca2+ response that remained elevated for 3 ± 0.05 times longer than in KNRK-PAR2 cells (Fig. 2 b). Homologous desensitization was assessed by repeated challenge of cells with AP and trypsin. Incubation of KNRK-PAR2 cells with 10 μM AP for 5 min desensitized the response to 10 nM trypsin, which was applied 5 min later, by 80 ± 10% compared with the vehicle-treated control cells (Fig. 2 c). In contrast, this treatment did not induce discernible desensitization in KNRK-PAR2 (δST363/6A) cells. Heterologous desensitization was assessed by pretreatment of cells with PDB to activate PKC. Preincubation of KNRK-PAR2 cells with 1 μM PDB for 20 min abolished responses to 10 nM trypsin (Fig. 2 d). However, this treatment had no effect on trypsin-induced Ca2+ mobilization in KNRK-PAR2(δST363/6A) cells. These data suggest that the mutant PAR2(δST363/6A) receptor is resistant to homologous and heterologous desensitization.
Figure 2.

PAR2-mediated Ca2+ mobilization in KNRK-PAR2 cells and KNRK-PAR2δ(ST363/6A) cells. (a) Concentration-response analysis for KNRK-PAR2 (•) and KNRK-PAR2 (δST363/6A) (▴) cells. (b–d) Each line shows [Ca2+]i for individual KNRK-PAR2 cells (left) and KNRK-PAR2(δST363/6A) cells (right). (b) Note that the response to trypsin is prolonged in KNRK-PAR2(δST363/6A) cells. (c) Homologous desensitization in cells pretreated with 10 μM AP for 5 min before addition of 10 nM trypsin. (d) Heterologous desensitization in cells pretreated with 1 μM PDB for 20 min before addition of 10 nM trypsin. Note that KNRK-PAR2(δST363/6A) cells are resistant to desensitization.
To assess PAR2 endocytosis, we incubated cells with 50 μM AP and quantified the surface Flag immunoreactivity by flow cytometry (Déry et al. 1999). In KNRK-PAR2 or KNRK-PAR2+ARR-GFP cells, AP stimulated rapid internalization of PAR2 (33 ± 12% and 31 ± 10% internalization at 5 min; 73 ± 13% and 71 ± 15% at 30 min, respectively; Fig. 3 a, •). In marked contrast, in KNRK-PAR2(δST363/6A) cells, there was no significant internalization even after 30 min (Fig. 3 a, ▴). Similarly, AP-induced endocytosis of PAR2 was markedly diminished in KNRK-PAR2+ARR319-418 cells (Fig. 3 a, ○). In hBRIE cells, AP also stimulated rapid internalization of PAR2 (70 ± 5% at 5 min; 90 ± 10% at 30 min; Fig. 3 a, □). Thus, the PAR2 expressed in KNRK cells and enterocytes undergoes agonist-induced endocytosis. Endocytosis is inhibited by the expression of dominant negative β-arrestin319-418, but is not affected by overexpression of wild-type β-arrestin. PAR2(δST363/6A) is highly resistant to agonist-induced endocytosis.
Figure 3.
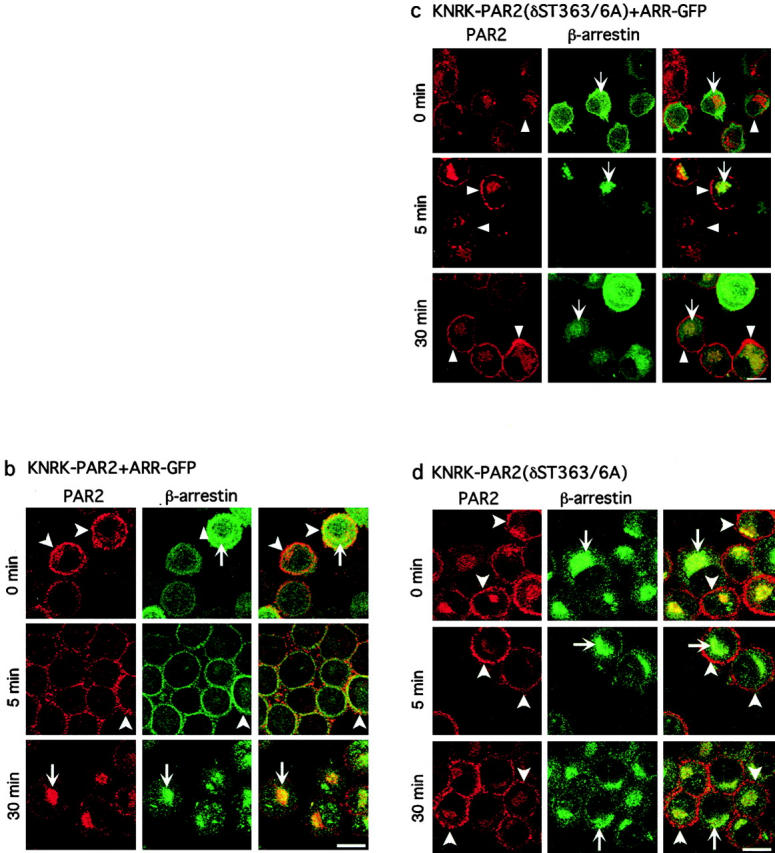

Agonist-induced PAR2 internalization. (a) Kinetics of PAR2 endocytosis in KNRK-PAR2+ARR-GFP cells (•), KNRK-PAR2 cells (⋄) KNRK-PAR2+ ARR319-418-GFP cells (○), KNRK-PAR2(δST363/6A) cells (▴), and hBRIE cells (□). Cells were incubated with 50 μM AP for 0–30 min at 37°C and endocytosis was determined by measuring surface Flag immunoreactivity by flow cytometry. *P < 0.05 compared with cells expressing PAR2 alone or PAR2 plus ARR-GFP cells, n = 3. (b–d) Localization of PAR2 and β-arrestin by immunofluorescence and confocal microscopy. KNRK-PAR2+ARR-GFP cells (b), KNRK-PAR2(δST363/6A)+ ARR-GFP cells (c), or KNRK-PAR2(δST363/6A) (d) were incubated with 50 nM trypsin for 0–30 min at 37°C. PAR2 was localized by immunofluorescence and β-arrestin was detected using GFP (b and c) or by immunofluorescence (d). The same cells are shown in each row and the images in the right panel are formed by superimposition of the images from the other two panels in the same row. Representative of two experiments. In KNRK-PAR2+ARR-GFP cells, note redistribution of β-arrestin to the plasma membrane at 5 min (arrowheads) and to endosomes at 30 min (arrows), where it colocalizes with PAR2. In KNRK-PAR2(δST363/6A)+ARR-GFP cells and in KNRK-PAR2(δST363/6A), note that PAR2 remains at the plasma membrane (arrowheads) and β-arrestin remains in the cytosol (arrows) with no colocalization. Bar, 10 μm.
To determine the effects of agonists on the subcellular distribution of PAR2 and β-arrestin, we incubated cells with 50 nM trypsin and localized PAR2 and β-arrestins by confocal microscopy. Agonist-induced trafficking of PAR2 is similar in KNRK-PAR2 cells and KNRK-PAR2+ARR-GFP cells (Böhm et al. 1996; Déry et al. 1999). In KNRK-PAR2+ARR-GFP cells, trypsin stimulated rapid redistribution of β-arrestin from the cytosol to the plasma membrane, where it colocalized with PAR2 at 5 min, followed by endocytosis of PAR2 and β-arrestins into the same endosomes at 30 min (Fig. 3 b; Déry et al. 1999). In KNRK-PAR2(δST363/6A) and KNRK-PAR2(δST363/6A)+ARR-GFP cells in the absence of agonist, PAR2 was present at the plasma membrane, ARR-GFP was diffusely localized in the cytosol and was sometimes detected at the plasma membrane, whereas immunoreactive endogenous β-arrestin was mostly detected in the cytosol (Fig. 3c and Fig. d). Trypsin did not induce endocytosis of PAR2(δST363/6A). ARR-GFP and endogenous β-arrestin were mostly retained in the cytosol and did not colocalize with PAR2 (δST363/6A) at the plasma membrane or in endosomes. These results indicate that PAR2(δST363/6A) does not interact with β-arrestins at the plasma membrane, which probably explains its resistance to desensitization and endocytosis. Furthermore, overexpression of ARR-GFP does not compensate for the inability of the mutant receptor to internalize.
We examined the ability of this internalization-resistant and desensitization-defective mutant receptor to activate ERK1/2. In KNRK-PAR2(δST363/6A) cells, 50 nM trypsin caused a 5.4 ± 1-fold increase in ERK activity at 5 min, which is similar to that observed in KNRK-PAR2 cells (Fig. 4 a). However, the ERK response, like the Ca2+ response, remained elevated in cells expressing PAR2 (δST363/6A). In KNRK-PAR2(δST363/6A) cells, trypsin stimulated a 6.1-fold increase in ERK1/2 activation (Fig. 4 b, ▴). Similar results were observed in HEK293 cells transfected with PAR2(δST363/6A) (not shown). Neither overexpression of ARR-GFP (Fig. 4 b, Δ) nor of ARR319-418-GFP (Fig. 4 b, ♦) in KNRK-PAR2(δST363/6A) cells altered ERK1/2 activation by the mutant receptor. Thus, whereas β-arrestin–dependent internalization of PAR2 is required for activation of ERK1/2 by the wild-type receptor, it is not required for a mutant receptor that is capable of prolonged signaling. These results suggest that the mutant receptor activates ERK1/2 by an alternative pathway that circumvents the requirement for receptor internalization.
Figure 4.

(a) PAR2-mediated ERK1/2 activity. KNRK-PAR2 (⋄) and KNRK-PAR2(δST363/6A) (▴) cells were treated with 50 nM trypsin for 0–30 min, and ERK activity was measured using the MBP assay. (b) Activation of ERK1/2. KNRK-PAR2(δST363/6A) (▴), KNRK-PAR2 (δST363/6A)+ARR-GFP (▵), and KNRK-PAR2 (δST363/6A)+ARR319-418-GFP (♦) cells were treated with 50 nM trypsin for 0–30 min and phosphorylation was assessed using antibodies to pERK1/2. *P < 0.05 compared with KNRK-PAR2 cells, n = 3.
Wild-type and Mutant PAR2 Activate ERK1/2 by Different Pathways
We compared the mechanism by which wild-type PAR2 and PAR2(δST363/6A) activate ERK1/2 to provide additional information about the importance of β-arrestins in agonist-induced activation of ERK1/2. Agonists of GPCRs can activate ERK1/2 by multiple pathways. For Gαi-coupled receptors, agonist-induced ERK activation is mediated by G protein βγ subunits and involves tyrosine phosphorylation of the same intermediates, such as shc and Grb2, used by receptor tyrosine kinases to activate ras and phosphorylate the MEK1 kinase raf-1, an event which is sensitive to pertussis toxin (Hawes et al. 1995; Luttrell et al. 1995; van Biesen et al. 1995; Daaka et al. 1997; Della Rocca et al. 1997, Della Rocca et al. 1999). GPCRs can also transactivate receptor tyrosine kinases (Luttrell et al. 1997a). In addition, both Ca2+ mobilization and EGF receptor transactivation can lead to PYK2 phosphorylation (Dikic et al. 1996; Lev et al. 1995; Eguchi et al. 1999; Zwick et al. 1999), which feeds into the ras-dependent pathway at the level of shc phosphorylation. Thus, there are a number of potential pathways by which PAR2 could activate ERK1/2.
Because activation of PAR2 leads to Ca2+ mobilization and stimulation of PKC, which can directly phosphorylate raf-1 (Hawes et al. 1995; Schönwasser et al. 1998), we first examined the role of PKC in PAR2-mediated activation of ERK1/2. We examined the effect a nonselective PKC inhibitor (GF109203X) and of a specific inhibitor of the Ca2+-dependent PKCβ (LY379196) on trypsin-stimulated ERK phosphorylation. In KNRK-PAR2 cells, both GF109203X and LY379196 reduced trypsin-stimulated ERK phosphorylation by 52 ± 5% and 48 ± 4%, respectively (Fig. 5 a). Likewise, in hBRIE cells, both GF109203X and LY379196 reduced trypsin-stimulated ERK1/2 phosphorylation by 78 ± 2% and 50 ± 5%, respectively (Fig. 5 a). In HEK293-PAR2 cells, GF109203X reduced trypsin-stimulated ERK1/2 phosphorylation by 70 ± 10% (not shown). However, in KNRK-PAR2+ARR319-418-GFP and KNRK-PAR2(δST363/6A) cells (Fig. 5 a), ERK1/2 activation was insensitive to both PKC inhibitors. Therefore, trypsin-stimulated activation of ERK1/2 by the wild-type receptor is predominantly mediated by PKC, some of which is through the specific PKCβ isozyme, whereas the mutant receptor activates ERK1/2 by a PKC-independent mechanism. To exclude the possibility that activation by PAR2 involves uncoupling from Gαq and coupling to a PTX-sensitive G protein, similar to β2-AR which uncouples from Gαs and couples to Gαi (Daaka et al. 1997), we examined the sensitivity of trypsin-stimulated ERK phosphorylation to PTX. Pretreatment with PTX did not affect trypsin-stimulated phosphorylation of ERK1/2 in KNRK-PAR2 cells (Fig. 5 a). If the internalization-defective PAR2(δST363/6A) activates ERK1/2 by a PKC and PTX-independent pathway, we hypothesized that it was doing so by a ras-dependent pathway involving phosphorylation of some of the same proteins used by receptor tyrosine kinases. To test this possibility, we examined the effect of the nonspecific tyrosine kinase inhibitor genistein, the epidermal growth factor receptor (EGFR) inhibitor tyrphostin 25, and the expression of dominant negative N17ras on trypsin-stimulated activation. In KNRK-PAR2(δST363/6A) cells, genistein abolished trypsin-stimulated activation of ERKs and tyrphostin 25–inhibited activation by 55 ± 5% (Fig. 5 a). In KNRK-PAR2 and hBRIE cells, both genistein and tyrphostin 25 inhibited trypsin activation of ERK1/2 by <20%. These results suggest that activation by the mutant receptor occurs via activation of tyrosine kinase pathways, in part, because of transactivation of EGFR, whereas the wild-type receptor activation of ERK1/2 is predominantly independent of tyrosine kinase activation. Expression of N17ras with PAR2(δST363/6A) but not wild-type PAR2 also abolished trypsin-stimulated activity, whereas ERK1/2 activation by 10% serum was abolished in both cell lines (Fig. 5 a). These results strongly suggest that the mutant receptor might utilize a ras-dependent pathway, whereas the wild-type receptor might not. However, it is somewhat difficult to interpret the effects of a dominant negative ras in a ras-transformed cell line. Unfortunately, attempts to express N17ras in either hBRIE or HEK293 cells have been unsuccessful. Therefore, to further clarify the difference between the mechanisms of ERK1/2 activation used by these two receptors, we examined other known components of the ras-dependent pathway.
Figure 5.

Mechanism of PAR2-mediated activation of ERK1/2. (a) KNRK-PAR2, hBRIE, KNRK-PAR2+ARR319-418, and KNRK-PAR2(δST363/6A) cells were untreated (control, con), or incubated with 100 nM GF109203X (GFX), 20 nM LY379196 (LY), 100 ng/ml PTX, 10 μM genistein (GEN), 20 μM tyrphostin 25 (TP), or were cotransfected with N17ras. Cells were incubated with 50 nM trypsin for 5 min. *P < 0.05 compared with untreated cells, n = 4. (b–f) Analysis of KNRK-PAR2 and KNRK-PAR2(δST363/6A) cells by immunoprecipitation (IP) and Western blotting (WB). Cells were incubated with 50 nM trypsin for 5 min. Extracts were immunoprecipitated with antibodies to PYK2 (b), Shc (c and d), and src (e–g). Blots were probed for phosphotyrosine (b–e), PYK2 (f), and src (g).
First, we examined tyrosine phosphorylation of PYK2 and the adaptor protein Shc. PYK2 is a member of the focal adhesion kinase family that has been shown to bind src, phosphorylate Shc, and activate ERK1/2 in response to extracellular signals that mobilize intracellular Ca2+, activate PKC, or transactivate EGFR (Dikic et al. 1996; Eguchi et al. 1999; Zwick et al. 1999). In KNRK-PAR2 cells, trypsin induced only a mild phosphorylation of PYK2 and shc (1.3 ± 0.1- and 1.2 ± 0.2-fold, respectively), compared with the more robust phosphorylation (3.7 ± 0.3- and 2.5 ± 0.2-fold, respectively) in KNRK-PAR2(δ363/6A) cells (Fig. 5b and Fig. c). This increase in PYK2 phosphorylation was similar to that observed for another Gαq-coupled receptor, the neurokinin-1 receptor (NK1R), activation of which results in nuclear ERK1/2 activity and proliferation (DeFea, K.A., and N.W. Bunnett, unpublished observations). In hBRIE cells, no significant trypsin-stimulated PYK2 phosphorylation (not shown) or shc phosphorylation was observed, although 10% of serum did stimulate shc phosphorylation (Fig. 5 d). Furthermore, upon trypsin treatment, tyrosine-phosphorylated PYK2 associated with src in KNRK-PAR2(δST363/6A) cells, whereas no significant increase in tyrosine-phosphorylated PYK2 association occurred in KNRK-PAR2 cells (Fig. 5 e), although a significant amount of unphosphorylated PYK2 was associated with src in unstimulated KNRK-PAR2 and KNRK-PAR2(δST363/6A) cells (Fig. 5 f).
Thus, β-arrestin–mediated receptor internalization is required for PAR2 activation of ERK1/2 only if the receptor is able to desensitize normally. If the signal generated by the receptor is prolonged because of point mutations, an alternate pathway is used. Wild-type PAR2 activates ERK1/2 principally by a β-arrestin– and PKC-dependent mechanism, whereas PAR2(δST363/6A) appears to couple to ERK1/2 by a tyrosine kinase– and ras-dependent mechanism. The mutant PAR2 pathway might also involve transactivation of EGFR, phosphorylation of PYK2 and shc, and activation of src.
PAR2 Association with β-Arrestins Determines the Localization and Specificity of ERK1/2
Our results raise the question of why the β-arrestin–dependent pathway for ERK1/2 activation exists. To address this question, we examined the next step in ERK1/2 activation, translocation of the activated kinases to the nucleus, to determine if the pathway of ERK1/2 activation affects the subcellular localization of the activated kinases.
To investigate whether activated ERK1/2 translocates to the nucleus, we incubated KNRK-PAR2 or KNRK-PAR2(δST363/6A) cells with 50 nM trypsin, 50 μM AP or 10% serum, and analyzed cytosolic and nuclear fractions for pERK immunoreactivity. In the cytosol, trypsin induced a 6 ± 2-fold increase in ERK1/2 phosphorylation in KNRK-PAR2 cells (Fig. 6 a, •) and a 7 ± 1-fold increase in KNRK-PAR2(δST363/6A) cells (Fig. 6 a, ▴) that was maintained for 5–30 min. In the nucleus, trypsin stimulated a large and rapid increase in ERK1/2 phosphorylation (14 ± 2-fold at 5 min) in KNRK-PAR2(δST363/6A) cells (Fig. 6 b, ▴). In marked contrast, trypsin stimulated only a minor increase in nuclear ERK1/2 phosphorylation in KNRK-PAR2 cells (Fig. 6 b, •). The minor increase in ERK1/2 activation observed in PAR2+ARR319-418-GFP cells appears in both nuclear and cytosolic fractions (Fig. 6, a and b, ○). However, the nuclear ERK1/2 activity observed in these cells was not significantly higher than that observed in KNRK-PAR2 cells. Similar results were obtained using AP (not shown). Addition of 10% serum stimulated a 10 ± 1.0-fold and a 14.6 ± 2-fold increase in cytosolic and nuclear ERK1/2 activation in both cell lines, respectively (not shown), which is similar to the nuclear activity observed with the mutant receptor. To ensure that cytosolic retention of ERK1/2 was not a result of exogenous PAR2 expression, we examined the subcellular localization of activated ERK1/2 in hBRIE cells. While treatment of hBRIE cells with 50 nM trypsin resulted in a 5.0 ± 1.0-fold increase in cytosolic ERK1/2 activity (Fig. 6 c, □), no marked increase in nuclear ERK1/2 activity was observed (Fig. 6 d, □). In contrast, 10% serum stimulated a 7.5 ± 1.5-fold increase in cytosolic and a 6.8 ± 0.8-fold increase in nuclear ERK1/2 activation (Fig. 6c and Fig. d, ♦).
Figure 6.

Nuclear translocation of ERK1/2. (a–d) Subcellular fractionation of activated ERK1/2. KNRK-PAR2+ARR-GFP cells (•), KNRK-PAR2+ ARR319-418-GFP cells (○), or PAR2(δST363/6A) cells (s) were incubated with 50 nM trypsin for 0–30 min at 37°C, and pERK was determined in the cytosolic (a) and nuclear (b) fractions (n = 3). hBRIE cells were incubated with 50 nM trypsin (□) or 10% serum (♦), and pERK was determined in the cytosolic (c) and nuclear (d) fractions. (e) KNRK-PAR2 and KNRK-PAR2(δST363/6A) cells, transiently transfected with ERK2-GFP, were incubated with 50 nM trypsin at 37°C, and translocation of ERK2-GFP was observed by confocal imaging. Representative of eight experiments. In KNRK-PAR2 cells, note that ERK2-GFP remains cytosolic but, in KNRK-PAR2(δST363/6A) cells, it redistributes to the nucleus. (f and g) Proliferative responses to AP and serum. KNRK-PAR2 (f, □) and KNRK-PAR2(δST363/6A) cells (f, ▪) or hBRIE cells (g) were incubated with 50 μM AP or 20% FCS for 24 h, and incorporation of [3H]thymidine and cell number were measured. *P < 0.05 compared with untreated cells or KNRK-PAR2 cells, n = 3.
To confirm these findings, that the mutant and wild-type PAR2 differ in their ability to stimulate nuclear translocation of activated ERKs, we examined trypsin-induced translocation of ERK2 tagged with GFP. We transiently expressed ERK2-GFP in KNRK-PAR2 or KNRK-PAR2(δST363/6A) cells, incubated cells with 50 nM trypsin, and observed cells by confocal microscopy. In KNRK-PAR2 cells, trypsin did not affect the distribution of ERK2-GFP, which remained in the cytosol at all time points (Fig. 6 e, left). In contrast, trypsin stimulated accumulation of ERK2-GFP in the nucleus of KNRK-PAR2 (δST363/6A) cells within 5 min, and it remained there for 30 min (Fig. 6 e, right). Thus, trypsin-stimulated ERK1/2 activity through the β-arrestin–dependent pathway is predominantly cytosolic.
Activation of the internalization-deficient PAR2 results in nuclear translocation of activated ERK1/2, an event that is a requirement for mitogenesis induced by a number of growth factors (Brunet et al. 1999). Therefore, we hypothesized that PAR2 agonists would stimulate proliferation of cells expressing PAR2(δST363/6A), but not in cells expressing wild-type PAR2 or in hBRIE cells. To assess proliferation, we incubated cells with 50 μM AP for 24 h and measured both [3H]thymidine incorporation into DNA and cell number. In KNRK-PAR2 cells, AP had no effect on [3H]thymidine incorporation or cell number (Fig. 6 f). In contrast, in KNRK-PAR2(δST363/6A) cells, AP induced a 2.4 ± 0.8-fold increase in [3H]thymidine incorporation and a 2.7 ± 0.5-fold increase in cell number, which is similar to that observed with serum. Similarly, in hBRIE cells, whereas serum induced a 2.5 ± 0.3-fold increase in [3H]thymidine incorporation and cell number, AP had no effect (Fig. 6 g).
Trypsin Induces the Formation of a Scaffolding Complex Containing PAR2, β-Arrestin, raf-1, and Activated ERK1/2
Disruption of the β-arrestin–dependent mechanism of ERK1/2 activation by receptor mutation led to the use of an alternative pathway that resulted in nuclear ERK1/2 activity and proliferation. Therefore, we hypothesized that β-arrestin is necessary for the cytosolic retention of the activated ERK1/2 through the formation of a scaffolding complex. Because previous studies of the β2-AR demonstrated agonist-induced association of β-arrestin and src (Luttrell et al. 1999), we examined the possibility that PAR2 agonists induce association of β-arrestin with components of the MAPK cascade. We incubated cells with 50 nM trypsin and localized β-arrestins and raf-1 by confocal microscopy. In KNRK-PAR2+ARR-GFP cells, trypsin stimulated translocation of β-arrestin and raf-1 from the cytosol to the plasma membrane, where they colocalized at 5 min (Fig. 7 a). Although trypsin also stimulated membrane translocation of raf-1 in cells expressing internalization-defective PAR2(δST363/6A)+ARR-GFP, β-arrestin remained in the cytosol and did not colocalize with raf-1 (Fig. 7 b). To confirm that colocalization of raf-1 and β-arrestin reflected protein–protein interaction, and to ensure that this association was not due to overexpression of ARR-GFP, KNRK-PAR2, and KNRK-PAR2(δST363/6A), cells were treated with 50 nM trypsin or 50 μM AP for 5 min, and the association of raf-1 with β-arrestin was determined by coimmunoprecipitation. While trypsin and AP stimulated a 5 ± 0.5- (Fig. 7 b) and 4.8 ± 0.8-fold increase (not shown), respectively, in association with β-arrestin and raf-1 over basal in KNRK-PAR2 cells, this association was not observed in KNRK-PAR2 (δST363/6A) cells (Fig. 7 c). Similar results were obtained in KNRK-PAR2+ARR-GFP cells (Fig. 7 d) and expression of ARR-GFP with PAR2(δST363/6A) did not compensate for the mutant receptor's inability to stimulate raf-1/β-arrestin association (Fig. 7 d). Expression of ARR319-418-GFP did not inhibit the ability of endogenous β-arrestin to bind raf-1, but the dominant negative β-arrestin did not associate with raf-1 (Fig. 7 e). In hBRIE+ ARR-GFP cells, trypsin induced a 2.8 ± 0.3-fold increase in raf-1 association with ARR-GFP after 5 min, and the association was maintained for 30 min (Fig. 7 f). Together, these data suggest that PAR2-mediated activation of ERK1/2 involves the formation of a signaling complex, which might serve to retain the activated ERK1/2 in the cytosol. To investigate this possibility, we incubated cells with 50 nM trypsin or 50 μM AP for 5 min, and fractionated lysates by gel filtration chromatography. Fractions within the included volume were analyzed by SDS-PAGE, and Western blots were probed for PAR2, β-arrestin, raf-1, pERK1/2, and total ERK1/2. For each of these proteins, the band density in every fraction is expressed as a percentage of the total protein detected in all fractions (see Materials and Methods) and complex formation is depicted in the graphs (Fig. 8). (Note that total ERK is represented as the sum of p-ERK and unphosphorylated ERK.) Representative Western blots of the fractions containing the complex are shown as insets. Since the expression of ARR-GFP does not increase ERK1/2 activation or raf-1/β-arrestin association, cells overexpressing ARR-GFP were not used in the gel filtration experiments.
Figure 7.
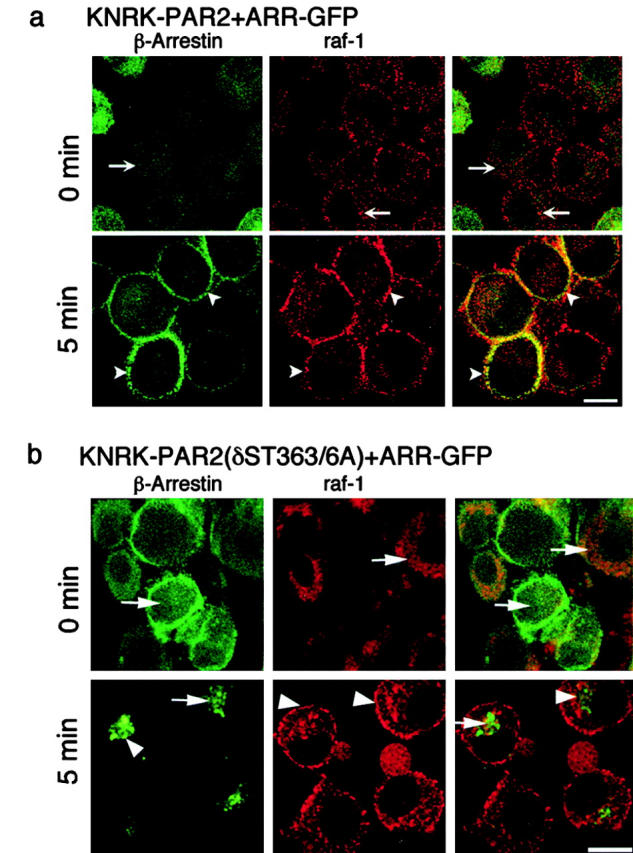

Trypsin induced association of β-arrestin and raf-1. (a and b) Localization of β-arrestin and raf-1 by immunofluorescence and confocal microscopy. KNRK-PAR2+ARR-GFP cells (a) or KNRK-PAR2(δST363/6A)+ ARR-GFP cells (b) were incubated with 50 nM trypsin for 0 or 5 min at 37°C. β-Arrestin was localized using GFP and raf-1 was localized by immunofluorescence. The same cells are shown in each row and the images in the right panel are formed by superimposition of images from the other two panels in the same row. Representative of two experiments. In KNRK-PAR2+ARR-GFP cells, note the redistribution of β-arrestin and raf-1 from the cytosol at 0 min (arrows) to the plasma membrane at 5 min (arrowheads), where they colocalize. In KNRK-PAR2 (δST363/6A)+ARR-GFP cells, note that β-arrestins remain in the cytosol (arrows) and raf-1 redistributes to the plasma membrane at 5 min (arrowheads). (c–f) Coimmunoprecipitation of raf-1 and β-arrestin. Cells were incubated with 50 nM trypsin for 0–30 min at 37°C, lysed, immunoprecipitated (IP) using antibodies to GFP or β-arrestin-1/2, and analyzed by Western blotting (WB) with a raf-1 antibody. (c) In KNRK-PAR2 cells, but not in KNRK-PAR2(δST363/6A) cells, β-arrestin and raf-1 coprecipitated. (d) Similarly, in KNRK-PAR2+ARR-GFP cells but not KNRK-PAR2(δST363/6A)+ARR-GFP cells ARR-GFP and raf-1 coprecipitated with antibodies to GFP. (e) In KNRK-PAR2+ARR319-418-GFP cells, endogenous β-arrestin and raf-1 coprecipitated, but ARR319-418 and raf-1 did not coprecipitate. (f) In hBRIE+ARR-GFP cells, ARR-GFP and raf-1 coprecipitated. Bars, 10 μm.
Figure 8.

Gel filtration analysis of an ERK signaling complex. KNRK-PAR2 (a and b), KNRK-PAR2(δST363/6A) cells (c and d), or hBRIE cells (e and f), or KNRK-PAR2+ARR319-418-GFP cells (g) were untreated (a, c, and e) or incubated with 50 μM AP (b, d, f, and g) for 5 min. Cell lysates were fractionated on a S300 Sephacryl column. The presence of pERK, raf-1, β-arrestin-1, and PAR2 in each fraction was determined by Western blotting (inset). PAR2 was detected using HA.11 antibody in KNRK cells and 2N antibody in hBRIE cells. Results are expressed as a percentage of the total protein for each partition coefficient (σ) (n = 3). The bracketed columns represent regions where proteins coeluted. Representative Western blots are shown of fractions containing the complex in KNRK-PAR2 cells (★, σ = 0.34), hBRIE cells (⋆, σ = 0.31–34), and in KNRK-PAR2+ARR319-418-GFP (*, σ = 0.45). (h) The Stoke's radii of the four molecular mass standards and the complex in KNRK-PAR2 cells (★, ∼6.2), hBRIE cells (⋆, ∼6.2–6.6), and KNRK-PAR2+ARR319-418-GFP (*, ∼5 nm) are graphed as a function of the error function complement of σ.
In KNRK-PAR2 and hBRIE cells, PAR2 agonists caused a marked redistribution in the elution profiles that resulted in the coelution of pERK, raf-1, β-arrestin, and PAR2 in the same fractions, with partition coefficients (σ) of 0.34 for KNRK-PAR2 cells and 0.31–0.34 for hBRIE cells (Fig. 8b and Fig. f, ★ and ⋆, respectively). In untreated cells (Fig. 8, a, c, and e) and in trypsin- or AP-stimulated KNRK-PAR2(δST363/6A) cells (Fig. 8 d), coelution of all four proteins was not observed. Notably, in trypsin- or AP-treated KNRK-PAR2 and hBRIE cells, >80% of the pERK coeluted with raf-1, β-arrestin, and PAR2. In contrast, in trypsin- or AP-treated KNRK-PAR2(δST363/6A) cells, <2% of the pERK eluted in these fractions. The approximate Stoke's radius of the putative complexes observed in stimulated KNRK-PAR2 and hBRIE cells was calculated from the error function complement of σ and is between 6 and 7 nm, which corresponds to a molecular mass of ∼250–300 kD (Fig. 8 h). Interestingly, in trypsin-stimulated KNRK-PAR2+ARR319-418-GFP cells, all four proteins coeluted at a partition coefficient of 0.45, although the ERK1/2 in the complex was not phosphorylated and the apparent Stoke's radius of the complex was only 5 nm. PAR2 activation resulted in coelution of PAR2, β-arrestin, raf-1, and pERK1/2 in KNRK-PAR2 and hBRIE cells. To determine if these proteins were associated in a complex, we used immunoprecipitation and Western blotting. Immunoprecipitation of gel filtration fractions from KNRK-PAR2 cells, containing the putative complex with antibodies to pERK1/2, coprecipitated PAR2, β-arrestin 1, and raf-1 (Fig. 9 a). Similarly, immunoprecipitation with antibodies to β-arrestin 1/2 coprecipitated pERK (Fig. 9 a). In hBRIE cells, antibodies to β-arrestin 1/2 coprecipitated pERK and raf-1 (Fig. 9 b). In both cell lines, anti-IgG did not precipitate PAR2, β-arrestin, raf-1, or pERK. Coprecipitation of PAR2 in hBRIE cells was hindered by the lack of PAR2 antibody sensitive enough to detect the low levels of protein that were purified in this complex. However, it is noteworthy that PAR2 coeluted in the same fractions as β-arrestin, raf-1, and pERK only upon trypsin or AP treatment, and that the size range of this putative complex is similar to that purified from transfected cells. From these data, we conclude that agonists of PAR2 induce the formation of a scaffolding complex near the inner leaflet of the plasma membrane, comprising internalized receptor, β-arrestin, raf-1, activated ERK, and possibly other members of the MAPK cascade.
Figure 9.

Coprecipitation of components of the β-arrestin–containing signaling complex. (a) Fractions from the gel filtration columns of AP-stimulated KNRK-PAR2 cells at partition coefficients 0.31–0.34 were pooled, concentrated, and immunoprecipitated with pERK or β-arrestin-1/2 antibodies. Western blots were probed for PAR2 using the HA.11 antibody, β-arrestin-1, raf-1, and pERK. (b) Fractions from the gel filtration columns of AP-stimulated hBRIE cells at partition coefficients 0.31–0.34 were similarly processed, immunoprecipitated with a β-arrestin-1/2 antibody, and blotted with antibodies to pERK and raf-1.
In KNRK-PAR2(δST363/6A) cells and untreated hBRIE cells, a portion of β-arrestin and phospho-ERK coeluted at partition coefficients 0.49–0.67. However, these proteins could not be coprecipitated (not shown) and probably do not associate. β-Arrestin might exist as a tetramer (Hirsch et al. 1999; Schubert et al. 1999), which could account for its faster migration than would be predicted for a protein of its size, although the significance of β-arrestin multimers is unclear. The coelution of small amounts of pERK, raf-1, and β-arrestin at partition coefficients >0.6 in untreated hBRIE cells (Fig. 8 e) cannot reflect a complex as the Stoke's radius of molecular mass standards in these fractions is <2.5 nm, the size of purified ERK alone (Khokhlatchev et al. 1998). The fact that endogenous β-arrestin can still associate with raf-1, and that these two proteins still coelute with unphosphorylated ERK1/2 and PAR2 even in the presence of ARR319-418, suggests that at least part of the complex, that comprising PAR2, β-arrestin, raf-1 and ERK1/2, assembles before interaction with the clathrin-coated pit. It will be of interest in the future to examine the effects of dominant negative mutants of proteins involved in later steps of endocytosis on ERK1/2 activation and complex assembly, and to identify other putative components of this complex.
Discussion
Although several studies have suggested a requirement for β-arrestin–mediated receptor endocytosis for agonist-induced ERK1/2 activity, the reason for this requirement has not been elucidated. Our results suggest that the Gαq-coupled receptor, PAR2, activates ERK1/2 through a PKC-dependent, PTX-insensitive pathway, by a mechanism that requires receptor association into a multiprotein complex comprising internalized receptor, β-arrestin, raf-1, activated ERK1/2, and perhaps other components of the MAPK pathway. Importantly, this complex forms in transfected cell lines and also in enterocytes that naturally express PAR2 and which are regulated under physiological circumstances by trypsin in the intestinal lumen (Kong et al. 1997). This complex might ensure that ERK1/2 activity remains in the cytosol and does not translocate to the nucleus to stimulate proliferation, and the data shown here support that hypothesis.
These findings suggest a novel role for scaffolding complexes in determining MAPK localization and the consequences of its activation (Fig. 10). A role for scaffolding complexes in the activation of ERK1/2 is an evolutionarily conserved one from yeast to mammalian cells (Luttrell et al. 1997b; Madhani et al. 1997). These scaffolding complexes bring components of the MAPK cascade in proximity to each other and, thereby, maintain substrate specificity. The PAR2/β-arrestin/raf-1/pERK complex may also serve to maintain substrate specificity, by preventing translocation of activated ERK1/2 to the nucleus, allowing phosphorylation only of cytosolic substrates. Comparisons between wild-type PAR2 and a mutant receptor, which does not interact with β-arrestins and thus fails to desensitize and internalize, provided important insights into the function of β-arrestin–dependent receptor trafficking. The mechanism by which an internalization and desensitization-defective mutant PAR2 activates ERK1/2 appears to involve activation of tyrosine kinases, possibly through the phosphorylation of PYK2, followed by its association with src and subsequent phosphorylation of the adaptor protein, Shc. The precise mechanism of ERK1/2 activation, however, has not been elucidated. Although both wild-type PAR2 and PAR2(δST363/6A) mobilize intracellular Ca2+, the prolonged signal exerted by the mutant receptor and its ability to transactivate the EGFR may account for its ability to activate PYK2, whereas the wild-type receptor does so only weakly (Fig. 10). The important point is that the mitogenic pathway available to the mutant receptor is unavailable to the wild-type receptor because ERK1/2 activation by PAR2 is tightly linked to β-arrestin–mediated desensitization and endocytosis. By preventing activated ERK1/2 from translocating to the nucleus, this mechanism may permit more precise control over a potentially mitogenic pathway.
Figure 10.

Model for β-arrestin–dependent and –independent activation of ERK1/2 by PAR2. (1a) Trypsin cleavage and activation of wild-type PAR2 leads to transient Ca2+ mobilization and activation of PKC. (1b) PKC phosphorylates and activates raf-1. (1c) PAR2 binds β-arrestin and desensitizes. (1d) Raf-1 and ERK1/2 associate with β-arrestin–bound receptor and (1e) the complex associates with the clathrin-coated pit and ERK becomes activated. (1f) Cytosolic, active ERK1/2 phosphorylate cytoskeletal proteins, microtubule-associated proteins (MAPs), and phospholipase A2 (PLA2). (2a) Trypsin cleavage and activation of PAR2(δST363/6A) leads to sustained Ca2+ mobilization and transactivation of EGFR. (2b) Tyrosine phosphorylation of kinases such as PYK2 occur, activating the classic ras-dependent ERK1/2 pathway. (2c) ERK1/2 translocate to the nucleus. (2d) ERK1/2 phosphorylate and activate nuclear proteins, leading to DNA replication and cell growth.
The mechanisms by which β-arrestin–containing complexes activate ERK1/2 and prevent their translocation to the nucleus remain to be determined. Peak ERK1/2 activity is observed after 5 min of trypsin stimulation, at which point only one third of the surface receptor is internalized, suggesting that either the only a portion of the PAR2, much of which is in the initial pool of internalized receptor, mediates the ERK1/2 response. The fact that dominant negative β-arrestin inhibits receptor association with clathrin-coated pits and ERK1/2 activation, but not association of ERK1/2 with β-arrestin–bound PAR2 and raf, suggests that early endosome formation might be required for activation of ERK1/2. Studies of purified ERK1/2 suggest that dimerization is a prerequisite for nuclear translocation (Khokhlatchev et al. 1998). Recent reports suggest that the association of MEK1 also serves to anchor unstimulated ERK1/2 in the cytoplasm of unstimulated cells (Fukuda et al. 1997) and that activated MEK is itself regulated by dynamin-mediated endocytosis (Kranenburg et al. 1999). In addition, a novel member of the importin family of nuclear transporters is required for nuclear translocation of p38, another member of the MAPK family (Ferrigno et al. 1998). Thus, the β-arrestin complex may sequester activated ERK1/2 to preclude their dimerization, prolong their association with MEK1, or impede their interaction with transport proteins, all of which could prevent nuclear translocation. In contrast, activation of the mutant PAR2 induced a shift in the apparent Stoke's radius of pERK (Fig. 8b and Fig. c), which would be consistent with the formation of ERK dimers, and with the ability of the mutant receptor to induce nuclear translocation of ERK1/2.
The β-arrestin–containing signaling complex is larger than might be predicted if only PAR2, β-arrestin, raf-1, and pERK were present at equal stoichiometries and, thus, the precise composition of the complex is presently unknown. We do not know whether pERK1/2 exists in the complex as monomers or dimers. Furthermore, the complex may contain other components of the MAPK pathway, such as MEK1, or other signaling proteins, such as PKC. Preliminary results suggest that MEK1 also coelutes from gel filtration columns with the complex described here, but it has been technically difficult to copurify as MEK1 with β-arrestin, perhaps because MEK1 dissociates from the complex during the experiment (DeFea, K.A., and N.W. Bunnett, unpublished observation). The association of the complex components identified here is quite stable, as it survives detergent solubilization, dilution, gel filtration, reconcentration, and immunoprecipitation; other complex components may be lost during these manipulations.
Nuclear translocation of ERK1/2 is an essential event in the control of gene activation and the cell growth and differentiation induced by some mitogens, and enforced cytoplasmic retention of activated ERK1/2 has been shown to inhibit growth factor–induced proliferation, while having no effect on phosphorylation of cytoplasmic substrates (Brunet et al. 1999). The cytoplasmic targets of PAR2-induced ERK1/2 remain to be determined. However, PAR2 agonists activate phospholipase A2 to generate arachidonic acid, and also stimulate cyclooxygenase-2 expression in an ERK-dependent manner in enterocytes (Kong, W., and N.W. Bunnett, unpublished observations). Cytosolic ERK1/2 may also phosphorylate cytoskeletal proteins, suggesting a role in cell migration and morphogenesis. In support of this possibility, preliminary studies in enterocytes suggest that PAR2 agonists stimulate formation of F-actin stress fibers in an ERK-dependent fashion (DeFea, K.A., and N.W. Bunnett, unpublished observations). Another potential target of cytosolic ERK is β-arrestin itself. Recent studies have shown that ERK phosphorylates β-arrestin, thereby impeding its ability to mediate endocytosis and MAPK signaling of the β2-AR (Lin et al. 1999). Thus, agonist-induced formation of this complex could bring ERK1/2 in proximity to β-arrestin, which would provide negative feedback control of receptor endocytosis. It remains to be determined whether association with β-arrestin also serves to cause cytoplasmic retention of ERK1/2, which are activated by agonists of other GPCRs. Furthermore, naturally occurring variants of GPCRs have been identified that exhibit diminished agonist-induced desensitization (Li et al. 1997) and internalization (Böhm et al. 1997), probably because of the inability to interact with β-arrestins. It will be of great interest to determine the mitogenic potential of these receptors and to investigate whether such variants of PAR2 also exist.
Acknowledgments
We thank D.K. Ways (Lily Inc., Indianapolis, IN) for LY379196, M. Cobb for rat-ERK2 plasmid, R.J. Lefkowitz for β-arrestin-1/2 antibody and β-arrestin-1 cDNA, J.H. Walsh for GFP antibody, M. Hollenberg for PAR2 antibody B5, P. Andrade-Gordon for PAR2 N antibody, G. Aponte for hBRIE 380 cells, A. Weiss for N17ras, R. Roth for HEK293 cells and P. Dazin for assistance with flow cytometry. We thank J. Ferrell (Stanford University, Stanford, CA) for careful review of this manuscript. We thank M. Lovett and E. O'Bryan (both from UCSF) for technical assistance.
This study was supported by National Institutes of Health grants DK43207, DK39957, and T32GM08258.
Footnotes
Abbreviations used in this paper: β2-AR, β2-adrenergic receptor; σ, partition coefficient; AP, activating peptide SLIGRL/KV-NH2; EGFP, enhanced green fluorescent protein; EGFR, epidermal growth factor receptor; ERK1/2, extracellular signal–regulated kinase 1 and 2; GPCR, G protein–coupled receptor; MAPK, mitogen-activated protein kinase; MBP, myelin basic protein; PAR2, proteinase-activated receptor; pERK, phosphorylated ERK; PDB, 4α-phorbol 12,13-dicanoate; PKC, protein kinase C; PTX, pertussis toxin.
References
- Alblas J., van Etten I., Moolenaar W.H. Truncated, desensitization-defective neurokinin receptors mediate sustained MAP kinase activation, cell growth and transformation by a Ras-independent mechanism. EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:3351–3360. [PMC free article] [PubMed] [Google Scholar]
- Böhm S.K., Khitin L., Smeekens S.P., Grady E.F., Payan D.G., Bunnett N.W. Identification of potential tyrosine-containing endocytic motifs in the carboxyl-tail and seventh transmembrane domain of the neurokinin 1 receptor. J. Biol. Chem. 1997;272:2363–2372. doi: 10.1074/jbc.272.4.2363. [DOI] [PubMed] [Google Scholar]
- Böhm S.K., Khitin L.M., Grady E.F., Aponte G., Payan D.G., Bunnett N.W. Mechanisms of desensitization and resensitization of proteinase-activated receptor-2. J. Biol. Chem. 1996;271:22003–22016. doi: 10.1074/jbc.271.36.22003. [DOI] [PubMed] [Google Scholar]
- Bornfeldt K.E., Campbell J.S., Koyama H., Argast G.M., Leslie C.C., Raines E.W., Krebs E.G., Ross R. The mitogen-activated protein kinase pathway can mediate growth inhibition and proliferation in smooth muscle cells. Dependence on the availability of downstream targets. J. Clin. Invest. 1997;100:875–885. doi: 10.1172/JCI119603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A., Roux D., Lenormand P., Dowd S., Keyse S., Pouysségur J. Nuclear translocation of p42/p44 mitogen-activated protein kinase is required for growth factor-induced gene expression and cell cycle entry. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:664–674. doi: 10.1093/emboj/18.3.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R.S., Sarnecki C., Blenis J. Nuclear localization and regulation of erk- and rsk-encoded protein kinases. Mol. Cell. Biol. 1992;12:915–927. doi: 10.1128/mcb.12.3.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun, P.K., SJ; Stanley, CA; Ackers, GK. 5-32. (UI: 69288391). 1969. Determination of the equilibrium constants of associating protein systems. 3. Evaluation of the weight fraction of monomer from the weight-average partition coefficient (application to bovine liver glutamate dehydrogenase). Biochemistry. 8:162. [DOI] [PubMed]
- Corvera C.U., Déry O., McConalogue K., Böhm S.K., Khitin L.M., Caughey G.H., Payan D.G., Bunnett N.W. Mast cell tryptase regulates rat colonic myocytes through proteinase-activated receptor 2. J. Clin. Invest. 1997;100:1383–1393. doi: 10.1172/JCI119658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corvera C.U., Déry O., McConalogue K., Gamp P., Thoma M., Al-Ani B., Caughey G.H., Hollenberg M.D., Bunnett N.W. Thrombin and mast cell tryptase regulate guinea-pig myenteric neurons through proteinase-activated receptors-1 and -2. J. Physiol. 1999;517:741–756. doi: 10.1111/j.1469-7793.1999.0741s.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daaka Y., Luttrell L.M., Lefkowitz R.J. Switching of the coupling of the beta2-adrenergic receptor to different G proteins by protein kinase A. Nature. 1997;390:88–91. doi: 10.1038/36362. [DOI] [PubMed] [Google Scholar]
- Daaka Y., Luttrell L.M., Ahn S., Della Rocca G.J., Ferguson S.S., Caron M.G., Lefkowitz R.J. Essential role for G protein-coupled receptor endocytosis in the activation of mitogen-activated protein kinase. J. Biol. Chem. 1998;273:685–688. doi: 10.1074/jbc.273.2.685. [DOI] [PubMed] [Google Scholar]
- DeFea K., Roth R.A. Modulation of insulin receptor substrate-1 tyrosine phosphorylation and function by mitogen-activated protein kinase. J. Biol. Chem. 1997;272:31400–31406. doi: 10.1074/jbc.272.50.31400. [DOI] [PubMed] [Google Scholar]
- Della Rocca G.J., van Biesen T., Daaka Y., Luttrell D.K., Luttrell L.M., Lefkowitz R.J. Ras-dependent mitogen-activated protein kinase activation by G protein-coupled receptors. Convergence of Gi- and Gq-mediated pathways on calcium/calmodulin, Pyk2, and Src kinase. J. Biol. Chem. 1997;272:19125–19132. doi: 10.1074/jbc.272.31.19125. [DOI] [PubMed] [Google Scholar]
- Della Rocca G.J., Maudsley S., Daaka Y., Lefkowitz R.J., Luttrell L.M. Pleiotropic coupling of G protein-coupled receptors to the mitogen-activated protein kinase cascade. Role of focal adhesions and receptor tyrosine kinases. J. Biol. Chem. 1999;274:13978–13984. doi: 10.1074/jbc.274.20.13978. [DOI] [PubMed] [Google Scholar]
- Déry O., Corvera C.U., Steinhoff M., Bunnett N.W. Proteinase-activated receptorsnovel mechanisms of signaling by serine proteases. Am. J. Physiol. 1998;274:C1429–C1452. doi: 10.1152/ajpcell.1998.274.6.C1429. [DOI] [PubMed] [Google Scholar]
- Déry O., Thoma M.S., Wong H., Grady E.F., Bunnett N.W. Trafficking of proteinase-activated receptor-2 and beta-arrestin-1 tagged with green fluorescent protein. beta-arrestin-dependent endocytosis of a proteinase receptor. J. Biol. Chem. 1999;274:18524–18535. doi: 10.1074/jbc.274.26.18524. [DOI] [PubMed] [Google Scholar]
- Dikic I., Tokiwa G., Lev S., Courtneidge S.A., Schlessinger J. A role for Pyk2 and Src in linking G-protein-coupled receptors with MAP kinase activation. Nature. 1996;383:547–550. doi: 10.1038/383547a0. [DOI] [PubMed] [Google Scholar]
- Eguchi S., Iwasaki H., Inagami T., Numaguchi K., Yamakawa T., Motley E.D., Owada K.M., Marumo F., Hirata Y. Involvement of PYK2 in angiotensin II signaling of vascular smooth muscle cells. Hypertension. 1999;33:201–206. doi: 10.1161/01.hyp.33.1.201. [DOI] [PubMed] [Google Scholar]
- Ferrigno P., Posas F., Koepp D., Saito H., Silver P.A. Regulated nucleo/cytoplasmic exchange of HOG1 MAPK requires the importin beta homologs NMD5 and XPO1. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:5606–5614. doi: 10.1093/emboj/17.19.5606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda M., Gotoh Y., Nishida E. Interaction of MAP kinase with MAP kinase kinaseits possible role in the control of nucleocytoplasmic transport of MAP kinase. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:1901–1908. doi: 10.1093/emboj/16.8.1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M., Jakes R., Spillantini M.G., Hasegawa M., Smith M.J., Crowther R.A. Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature. 1996;383:550–553. doi: 10.1038/383550a0. [DOI] [PubMed] [Google Scholar]
- Hawes B.E., van Biesen T., Koch W.J., Luttrell L.M., Lefkowitz R.J. Distinct pathways of Gi- and Gq-mediated mitogen-activated protein kinase activation. J. Biol. Chem. 1995;270:17148–17153. doi: 10.1074/jbc.270.29.17148. [DOI] [PubMed] [Google Scholar]
- Hirsch J.A., Schubert C., Gurevich V.V., Sigler P.B. The 2.8 A crystal structure of visual arrestina model for arrestin's regulation. Cell. 1999;97:257–269. doi: 10.1016/s0092-8674(00)80735-7. [DOI] [PubMed] [Google Scholar]
- Ignatova E.G., Belcheva M.M., Bohn L.M., Neuman M.C., Coscia C.J. Requirement of receptor internalization for opioid stimulation of mitogen-activated protein kinasebiochemical and immunofluorescence confocal microscopic evidence. J. Neurosci. 1999;19:56–63. doi: 10.1523/JNEUROSCI.19-01-00056.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khokhlatchev A.V., Canagarajah B., Wilsbacher J., Robinson M., Atkinson M., Goldsmith E., Cobb M.H. Phosphorylation of the MAP kinase ERK2 promotes its homodimerization and nuclear translocation. Cell. 1998;93:605–615. doi: 10.1016/s0092-8674(00)81189-7. [DOI] [PubMed] [Google Scholar]
- Klemke R.L., Cai S., Giannini A.L., Gallagher P.J., de Lanerolle P., Cheresh D.A. Regulation of cell motility by mitogen-activated protein kinase. J. Cell Biol. 1997;137:481–492. doi: 10.1083/jcb.137.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong W., McConalogue K., Khitin L.M., Hollenberg M.D., Payan D.G., Böhm S.K., Bunnett N.W. Luminal trypsin may regulate enterocytes through proteinase-activated receptor 2. Proc. Natl. Acad. Sci. USA. 1997;94:8884–8889. doi: 10.1073/pnas.94.16.8884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranenburg O., Verlaan I., Moolenaar W.H. Dynamin is required for the activation of mitogen-activated protein (MAP) kinase by MAP kinase kinase. J. Biol. Chem. 1999;274:35301–35304. doi: 10.1074/jbc.274.50.35301. [DOI] [PubMed] [Google Scholar]
- Lev S., Moreno H., Martinez R., Canoll P., Peles E., Musacchio J.M., Plowman G.D., Rudy B., Schlessinger J. Protein tyrosine kinase PYK2 involved in Ca(2+)-induced regulation of ion channel and MAP kinase functions. Nature. 1995;376:737–745. doi: 10.1038/376737a0. [DOI] [PubMed] [Google Scholar]
- Li H., Leeman S.E., Slack B.E., Hauser G., Saltsman W.S., Krause J.E., Blusztajn J.K., Boyd N.D. A substance P (neurokinin-1) receptor mutant carboxyl-terminally truncated to resemble a naturally occurring receptor isoform displays enhanced responsiveness and resistance to desensitization. Proc. Natl. Acad. Sci. USA. 1997;94:9475–9480. doi: 10.1073/pnas.94.17.9475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin F.T., Miller W.E., Luttrell L.M., Lefkowitz R.J. Feedback regulation of beta-arrestin1 function by extracellular signal-regulated kinases. J. Biol. Chem. 1999;274:15971–15974. doi: 10.1074/jbc.274.23.15971. [DOI] [PubMed] [Google Scholar]
- Luttrell L.M., van Biesen T., Hawes B.E., Koch W.J., Touhara K., Lefkowitz R.J. G betagamma subunits mediate mitogen-activated protein kinase activation by the tyrosine kinase insulin-like growth factor 1 receptor. J. Biol. Chem. 1995;270:16495–16498. doi: 10.1074/jbc.270.28.16495. [DOI] [PubMed] [Google Scholar]
- Luttrell L.M., Della Rocca G.J., van Biesen T., Luttrell D.K., Lefkowitz R.J. G betagamma subunits mediate Src-dependent phosphorylation of the epidermal growth factor receptor. A scaffold for G protein-coupled receptor-mediated Ras activation J. Biol. Chem 272 1997. 4637 4644a [DOI] [PubMed] [Google Scholar]
- Luttrell L.M., van Biesen T., Hawes B.E., Koch W.J., Krueger K.M., Touhara K., Lefkowitz R.J. G-protein-coupled receptors and their regulationactivation of the MAP kinase signaling pathway by G-protein-coupled receptors Adv. Second Messenger Phosphoprotein Res 31 1997. 263 277b [PubMed] [Google Scholar]
- Luttrell L.M., Ferguson S.S., Daaka Y., Miller W.E., Maudsley S., Della Rocca G.J., Lin F., Kawakatsu H., Owada K., Luttrell D.K., Caron M.G., Lefkowitz R.J. Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science. 1999;283:655–661. doi: 10.1126/science.283.5402.655. [DOI] [PubMed] [Google Scholar]
- Madhani H.D., Styles C.A., Fink G.R. MAP kinases with distinct inhibitory functions impart signaling specificity during yeast differentiation. Cell. 1997;91:673–684. doi: 10.1016/s0092-8674(00)80454-7. [DOI] [PubMed] [Google Scholar]
- McConalogue K., Corvera C.U., Gamp P.D., Grady E.F., Bunnett N.W. Desensitization of the neurokinin-1 receptor (NK1-R) in neuronseffects of substance P on the distribution of NK1-R, Galphaq/11, G-protein receptor kinase-2/3, and beta-arrestin-1/2. Mol. Biol. Cell. 1998;9:2305–2324. doi: 10.1091/mbc.9.8.2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T.D., Moody M.W., Steinhoff M., Okolo C., Koh D.S., Bunnett N.W. Trypsin activates pancreatic duct epithelial cell ion channels through proteinase-activated receptor-2. J. Clin. Invest. 1999;103:261–269. doi: 10.1172/JCI2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Post G.R., Collins L.R., Kennedy E.D., Moskowitz S.A., Aragay A.M., Goldstein D., Brown J.H. Coupling of the thrombin receptor to G12 may account for selective effects of thrombin on gene expression and DNA synthesis in 1321N1 astrocytoma cells. Mol. Biol. Cell. 1996;7:1679–1690. doi: 10.1091/mbc.7.11.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouyssegur J., Böhm S.K. Transmembrane receptors and intracellular pathways that control cell proliferation. Annu. Rev. Physiol. 1992;54:195–210. doi: 10.1146/annurev.ph.54.030192.001211. [DOI] [PubMed] [Google Scholar]
- Ray L.B., Sturgill T.W. Characterization of insulin-stimulated microtubule-associated protein kinase. Rapid isolation and stabilization of a novel serine/threonine kinase from 3T3-L1 cells. J. Biol. Chem. 1988;263:12721–12727. [PubMed] [Google Scholar]
- Schönwasser D.C., Marais R.M., Marshall C.J., Parker P.J. Activation of the mitogen-activated protein kinase/extracellular signal-regulated kinase pathway by conventional, novel, and atypical protein kinase C isotypes. Mol. Cell. Biol. 1998;18:790–798. doi: 10.1128/mcb.18.2.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert C., Hirsch J.A., Gurevich V.V., Engelman D.M., Sigler P.B., Fleming K.G. Visual arrestin activity may be regulated by self-association. J. Biol. Chem. 1999;274:21186–21190. doi: 10.1074/jbc.274.30.21186. [DOI] [PubMed] [Google Scholar]
- Sturgill T., Ray L.B. Muscle proteins related to microtubule associated protein-2 are substrates for an insulin-stimulatable kinase. Biochem. Biophys. Res. Commun. 1986;29:565–571. doi: 10.1016/s0006-291x(86)80457-0. [DOI] [PubMed] [Google Scholar]
- van Biesen T., Hawes B.E., Luttrell D.K., Krueger K.M., Touhara K., Porfiri E., Sakaue M., Luttrell L.M., Lefkowitz R.J. Receptor-tyrosine-kinase- and G beta gamma-mediated MAP kinase activation by a common signalling pathway. Nature. 1995;376:781–784. doi: 10.1038/376781a0. [DOI] [PubMed] [Google Scholar]
- Vögler O., Nolte B., Voss M., Schmidt M., Jakobs K.H., van Koppen C.J. Regulation of muscarinic acetylcholine receptor sequestration and function by beta-arrestin. J. Biol. Chem. 1999;274:12333–12338. doi: 10.1074/jbc.274.18.12333. [DOI] [PubMed] [Google Scholar]
- Zwick E., Wallasch C., Daub H., Ullrich A. Distinct calcium-dependent pathways of epidermal growth factor receptor transactivation and PYK2 tyrosine phosphorylation in PC12 cells. J. Biol. Chem. 1999;274:20989–20996. doi: 10.1074/jbc.274.30.20989. [DOI] [PubMed] [Google Scholar]