

Abstract
We performed a multitiered, case-control association study of psoriasis in three independent sample sets of white North American individuals (1,446 cases and 1,432 controls) with 25,215 genecentric single-nucleotide polymorphisms (SNPs) and found a highly significant association with an IL12B 3′-untranslated-region SNP (rs3212227), confirming the results of a small Japanese study. This SNP was significant in all three sample sets (odds ratio [OR]common 0.64, combined P [Pcomb]=7.85×10-10). A Monte Carlo simulation to address multiple testing suggests that this association is not a type I error. The coding regions of IL12B were resequenced in 96 individuals with psoriasis, and 30 additional IL12B-region SNPs were genotyped. Haplotypes were estimated, and genotype-conditioned analyses identified a second risk allele (rs6887695) located ∼60 kb upstream of the IL12B coding region that exhibited association with psoriasis after adjustment for rs3212227. Together, these two SNPs mark a common IL12B risk haplotype (ORcommon 1.40, Pcomb=8.11×10-9) and a less frequent protective haplotype (ORcommon 0.58, Pcomb=5.65×10-12), which were statistically significant in all three studies. Since IL12B encodes the common IL-12p40 subunit of IL-12 and IL-23, we individually genotyped 17 SNPs in the genes encoding the other chains of these cytokines (IL12A and IL23A) and their receptors (IL12RB1, IL12RB2, and IL23R). Haplotype analyses identified two IL23R missense SNPs that together mark a common psoriasis-associated haplotype in all three studies (ORcommon 1.44, Pcomb=3.13×10-6). Individuals homozygous for both the IL12B and the IL23R predisposing haplotypes have an increased risk of disease (ORcommon 1.66, Pcomb=1.33×10-8). These data, and the previous observation that administration of an antibody specific for the IL-12p40 subunit to patients with psoriasis is highly efficacious, suggest that these genes play a fundamental role in psoriasis pathogenesis.
Psoriasis is a common, chronic, T-cell–mediated inflammatory disease of the skin, affecting ∼2%–3% of whites of European descent. Although this disease is found in all populations, its prevalence is lower in Asians and African Americans and also declines at lower latitudes.1 The most common form, psoriasis vulgaris, is characterized by varying numbers of red, raised, scaly skin patches that can be present on any body surface but that most often appear on the elbows, knees, and scalp. The onset of disease usually occurs early in life (ages 15–30 years) and affects males and females equally. Up to 30% of individuals with psoriasis will develop an inflammatory arthritis, which can affect the peripheral joints of the hands and feet, large joints, or the central axial skeleton.2,3 Pathologically, psoriasis is characterized by vascular changes, hyperproliferation of keratinocytes, altered epidermal differentiation, and inflammation.4 In particular, the reaction of cells in the epidermis to type 1 effector molecules produced by T-cells results in the characteristic pathology of the plaques.5
The genetics of psoriasis are complex, and this disease is highly heritable, as evidenced by an increased rate of concordance in MZ versus DZ twins (35%–72% vs. 12%–23%, respectively) and a substantially increased incidence in family members of affected individuals (e.g., 6% for first-degree relatives); however, it is clear that environmental effects are also responsible for disease susceptibility.5 Ten genomewide linkage scans have resulted in strong evidence of a susceptibility locus (PSORS1 [MIM #177900]) in the major histocompatibility complex (MHC) on 6p21 but have not yielded consistent evidence for other regions (for a review, see the work of Bowcock and Kreuger5).
Linkage and association in the MHC (6p21) are thought to be due to human leukocyte antigen (HLA)–C. In particular, psoriasis-susceptibility effects are thought to be caused by the *0602 allele,6,7 although other candidate genes in the region may also contribute to disease predisposition. Association studies have identified three gene regions under linkage peaks with considerable evidence of linkage disequilibrium (LD) with psoriasis—namely, SLC9A3R1/NAT9 and RAPTOR (KIAA1303) in 17q25 and SLC12A8 in 3q21.8,9 Several other genes—including IL12B (MIM 161561), VDR, MMP2, IL10, IL1RN, and IRF2 (Genetic Association Database)—have been associated with psoriasis in sample sets of varying sizes and of different ethnicities; however, without more data from additional independent studies, it is difficult to draw statistically sound conclusions about whether or not they are true disease genes.
Genomewide association studies have been promoted as a powerful method for the mapping of complex traits, and both direct (i.e., with use of protein-altering markers) and indirect (i.e., with tag SNPs or spaced markers) strategies have been proposed.10–12 We undertook a large, genecentric association study that focused primarily on putative functional SNPs, to identify psoriasis-susceptibility markers in white North American case-control samples, and attempted to replicate significant markers in a second independent case-control sample set. During this process, we observed significant association of a SNP (rs3212227) in the 3′ UTR of IL12B with psoriasis in both case-control sample sets, confirming the results observed in a single Japanese study.13
To explore the role of IL12B in the genetics of psoriasis and to identify potentially causative variants, we genotyped additional markers in and around IL12B and performed further analyses, including haplotype analyses. Because IL12B encodes the IL-12p40 subunit of two distinct heterodimeric cytokines—IL-12 and IL-2314—we analyzed SNPs in IL12A, IL23A (encoding the IL-12p35 and IL-23p19 subunits, respectively), and the genes encoding the receptors of IL-12 and IL-23 (IL12RB1, IL12RB2, and IL23R [MIM 607562]), to determine if polymorphisms within these IL12B-related genes also play a role in psoriasis susceptibility. Finally, we replicated all interesting findings in a third independent white North American sample set.
Subjects and Methods
Overall Strategy
We conducted three sequential case-control studies (i.e., discovery, replication 1, and replication 2), to identify SNPs associated with psoriasis. In the discovery study, DNA samples from individuals with (cases) and without (controls) psoriasis were genotyped for 25,215 SNPs, with the use of disease-phenotype–based pooled DNA samples, to increase genotyping throughput and to minimize DNA consumption. The allele frequency of each SNP was determined as described elsewhere,15 and SNPs associated with psoriasis at the .05 significance level were evaluated in an independent sample set (replication 1 study), by use of a similar pooling strategy. Non–MHC-linked SNPs that were associated with psoriasis in the replication 1 study (P<.05) and had the same risk allele as in the discovery study were then individually genotyped in both the discovery and the replication 1 sample sets, to verify the results from the pooled DNA studies. SNPs that were still significant (P<.05) were then interrogated in a third independent sample set (replication 2 study). Here, we report the results of the most significant non-MHC SNP.
Subjects
The discovery samples consisted of 467 white individuals with psoriasis and 500 white individuals who served as controls. All subjects are of northern European ancestry and reside in Utah or in adjacent regions of southern Idaho, the area served by the University of Utah Hospital and Clinics. The individuals with psoriasis were recruited from the University of Utah Department of Dermatology clinics, as part of the Utah Psoriasis Initiative (UPI), and had a diagnosis of psoriasis confirmed by a UPI investigator. A full clinical workup and a questionnaire were available for each individual with psoriasis, and samples from first- or second-degree relatives were not included in the study. Samples from 500 white individuals matched for sex and ethnicity were selected from the CEPH Utah (CEU) population (n=112 grandparents) and from other University of Utah research studies (n=388) to serve as controls. The other research studies include those of colon cancer, restless legs, smoking addiction, epilepsy, and retinal disorders, and all control subjects used in the study described here were either control subjects in these other research studies or were unrelated spouses from family studies. The 112 control subjects from the CEU population were ascertained without regard to disease status. For the remaining 388 individuals, information about autoimmune disease status was available from 53%, and any individual with psoriasis or another autoimmune disease was excluded. Information about autoimmune disease status was not available for the remaining 47%; however, we expect the frequency of psoriasis and other autoimmune diseases in this control population to be similar to that of the general Utah population. Note that, if any of these control subjects carry, with increased frequency, alleles for autoimmune diseases that are also found for psoriasis, the significance of the results reported here would increase if those controls were removed.
The replication 1 sample set was collected by the Genomics Collaborative Division of SeraCare Life Sciences (GCI). Samples were included from 498 unrelated white North American individuals with a dermatologist-confirmed diagnosis of psoriasis and a Psoriasis Area and Severity Index (PASI) score and from 498 unrelated white North American individuals without a history of psoriasis or other autoimmune disorders and individually matched to the cases by sex and age. The replication 2 sample set, consisting of samples from 481 unrelated white North American individuals with a dermatologist-confirmed diagnosis of psoriasis and from 424 unrelated white North Americans without psoriasis, was used to confirm results from the two previous sample sets; samples from 293 individuals with psoriasis and 292 individuals without psoriasis were provided by GCI and met the criteria described above. BioCollections Worldwide provided an additional 188 samples from individuals with psoriasis who met the same criteria described above, as well as samples from 132 white North Americans without psoriasis.
All individuals included in this study were 18 years or older at the time they were enrolled in the sample collections. In addition, none of the subjects had undergone a bone-marrow transplant. All protocols were approved by national and/or local institutional review boards, and informed written consent was obtained from all subjects. A detailed breakdown of the clinical characteristics of the discovery and replication 1 sample sets is provided in table 1. Detailed clinical information for all individuals with psoriasis in the replication 2 sample set was limited. The female:male sex ratio was 0.89, and the average (±SD) age of onset was 28 ± 16 years.
Table 1. .
Demographic and Clinical Information
| Sample Set |
||
| Subphenotypea | Discovery | Replication 1 |
| Genetic background | White (Utah) | White (North America) |
| No. of cases | 467 | 498 |
| No. of controls | 500 | 498 |
| Female:male sex ratio | 1.2 | 1.2 |
| Average age at onset (years) | 28 ± 17 | 29 ± 15 |
| Percentage with family history of psoriasis | 62.70% | 46.60% |
| No. of cases with: | ||
| Psoriatic arthritis and psoriasis for at least 10 years (yes/no) | 125/239 | 98/143 |
| National Psoriasis Foundation score <7.5/7.5–12.5/>12.5 | 124/132/129 | … |
| PASI score <10/10–50/>50 | … | 364/103/4 |
Data for certain subphenotypes were not available for all patients.
Scan SNP Selection
Allele-specific, kinetic PCR assays were developed for a collection of 25,215 genecentric SNPs curated from dbSNP, the Applera Genome Initiative,16,17 and the literature. SNPs were selected for inclusion if they appeared in more than one database and had a minor-allele frequency (MAF) ⩾1%. Of the SNPs, ∼70% are missense polymorphisms predicted to modify the amino acid sequences encoded by the genes. The majority of the remaining 30% of SNPs were splice-site acceptors or donors, putative transcription-factor binding sites, etc. Of the SNPs, ∼75% have MAFs ⩾5%.
Allele Frequency and Genotype Determination
SNP allele frequencies in pooled DNA samples were determined by allele-specific, kinetic PCR, as described elsewhere.15,16 In brief, 3 ng of pooled DNA was amplified using allele-specific primers and allele frequencies calculated from the two allele-specific PCR amplification curves, each determined in duplicate. Individual genotyping was performed using allele-specific kinetic PCR on 0.3 ng of DNA, and the data were hand curated before statistical analysis, without knowledge of case-control status. Genotyping calls were made on >99% of samples, for all SNPs genotyped. Previous analyses suggest a genotyping accuracy of >99%.18,19 Genotyping of the four most interesting SNPs described here (i.e., rs3212227, rs6887695, rs7530311, and rs11209026) in 320 of the psoriasis study samples, with use of an unrelated technology—a multiplexed oligoligation assay—showed >99.8% concordance. HLA-C genotypes were determined for the discovery samples by high-resolution sequence-based typing (performed by Atria Genetics). HLA-C genotypes were not available for the other two sample sets.
Pools were constructed using an orthogonal design in which an individual DNA sample was arrayed into multiple pools on the basis of clinically relevant phenotypes. For the unstratified analyses (i.e., all cases vs. all controls), the allele-frequency measurements in each stratum were combined and averaged on the basis of the formula
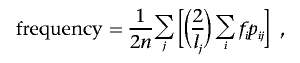 |
where lj is the number of pools in which sample j appears, pij=1 if sample j belongs to pool i and is 0 otherwise, fi is the allele-frequency estimate in pool i, and n is the number of distinct samples across all pools. By using an orthogonal pooling strategy based on clinically relevant phenotypes and by collapsing all the strata into an analysis of all cases versus all controls, we were essentially taking repeated measurements and were able to reduce measurement error for the comparison of all cases versus all controls.
IL12B Sequencing
To identify novel variants of IL12B, we selected DNA for resequencing from 96 randomly chosen individuals with psoriasis in the replication 1 sample set. Sequence data from all eight annotated exons and from the 5,000 bp upstream of the 5′-most exon of IL12B, which spans ∼17 kb, were extracted from the R27 draft of the Celera human genome sequence (Entrez Nucleotide [accession number NW_922784]). Primers were designed using the Primer3 program20 and included the M13 forward (5′ primer) or reverse (3′ primer) universal sequencing primer-binding sequence at their 5′ ends. PCR amplification and sequencing were performed as described elsewhere.19
SNP Selection for Follow-Up Genotyping
Tag SNPs for follow-up genotyping were selected by downloading the CEU HapMap genotypes between positions 158,464,177 and 158,828,966 on chromosome 5 from the HapMap Web site (release 12, October 2004) and by running the computer program Redigo.21 The approach used by this program maximizes the statistical power to detect truly associated SNPs, while minimizing the number of SNPs that are genotyped, and does not use measures of LD to select the SNPs. For the power calculations, we set the following parameters: 500 cases, 500 controls, 0.95 power threshold, disease prevalence 0.01, and a conservative disease model (additive mode of inheritance with genotypic relative risk [GRR] 1.5). As described by Hu and colleagues,21 use of a conservative disease model produces a tag SNP set that can exhibit moderate-to-substantial LD between pairs of selected SNPs and is also robust to changes in disease models.
LD Analysis and Haplotype Estimation
The LD measure r2 was calculated from unphased data with use of the LDMAX program in the GOLD package.22 Spotfire software was used to generate graphical representations of the r2 LD matrix. A pseudo-Gibbs sampling algorithm from the program SNPAnalyzer version 1.023 was used to estimate haplotype and diplotype frequencies from unphased data. Cases and controls were treated separately in this process. The Haplo.Stats package, which calculates global P values (P values that test the null hypothesis of independence between disease status and all the haplotypes) as well as P values for each individual predicted haplotype, was used to test for association between haplotypes and disease status.24 The Haplo.Stats package is particularly useful for unphased data, since it adjusts the variance on the test statistic for haplotype estimation error.
Association Statistics
A test of independence between allelic counts and disease status was performed using Fisher’s exact test.25 For larger contingency tables, the Williams-corrected G test was applied, to evaluate the null hypothesis of independence between genotype counts and psoriasis status,26 except when the data were sparse. In that situation, a Monte Carlo simulation approach was used to obtain appropriate P values. To explore the extent of deviation from Hardy-Weinberg equilibrium (HWE), Weir’s exact test for HWE was calculated.27 Since many disease models will predict a difference in pairwise LD between samples from affected individuals and samples from unaffected individuals, we used the LD contrast test, described by Nielsen and colleagues,28 to test for differences in LD patterns between cases and controls. We employed Fisher’s combined P value to perform a joint analysis across independent studies. A Mantel-Haenszel common odds ratio (OR) was calculated across independent studies.26 GRR estimates (denoted as γ) were calculated using a Bayesian approach modeling (i) the prevalence of disease as a beta distribution having an expected disease prevalence of 2.3% and (ii) the genotype counts as multinomially distributed with parameters estimated from the empirical data. The Breslow-Day statistic29 was used to determine whether SNPs exhibited significant heterogeneity of ORs across subphenotypes. Estimates of the population-attributable fraction were calculated as described by Walter30 and Schlesselman.31
Since false-positive results can be problematic in any large-scale experiment in which modest nominal significance levels are used, we performed a Monte Carlo simulation to obtain the distribution of combined P values across the two studies. Two null hypotheses were considered, and the results were presented under each hypothesis. Under the first model, which assumed independence between the cases and controls and a uniform distribution of P values, we generated 26,000 stochastically independent P values, retaining the top 1,500 most significant. A second set of 1,500 P values were then generated under a uniform distribution, to simulate P values for the replication data set, and the two P values for each replicate were combined using Fisher’s combined P value. The entire in silico experiment was repeated 1,000 times. The single most significant P value from each in silico experiment was then used to generate a distribution of the 1,000 most significant experimentwide P values. This is analogous to results obtained under the Dunn-Sidak–corrected significance level (similar to a Bonferroni correction).26 For the second model, we took a similar approach, except that we used the results from the pooled discovery samples to model the distribution of null P values. This approach is typically more conservative and is described in detail by Schrodi.32 The resulting gamma-null model was defined by scale and shape parameters of 1.2032 and 0.8517, respectively. Similar to other empirically based methods used in large-scale studies to obtain null or alternative models,33 this gamma-model method concurrently adjusts for diffuse effects of population stratification in a way similar to genomic control approaches.34
In an approach similar to the haplotype method,35,36 we performed tests of association for a specific SNP conditional on genotypes at other SNPs. That is, cases and controls were both partitioned on the basis of genotypes at one SNP, and the counts at an interrogated SNP were used in a test statistic. The statistical significance of the resulting test statistic was assessed using a permutation procedure. This type of test is useful in dissecting the relative contribution of each SNP to disease association for a set of SNPs in LD.
To explore the likelihood of our data under various disease models, we calculated Bayes factor curves,37,38 using data from all three studies. The results were compared against the likelihood of the data under a null hypothesis that accounts for diffuse population stratification. Mathematically, the Bayes factor can be written as 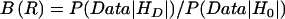 , where R is the allelic relative risk, HD is the disease model, which is a function of R, and H0 is the null hypothesis.32
, where R is the allelic relative risk, HD is the disease model, which is a function of R, and H0 is the null hypothesis.32
Results
Discovery Scan and Replication Data
A genecentric set of 25,215 SNPs was genotyped in pooled discovery samples consisting of 466 white North American individuals with psoriasis and 500 white North American control individuals from Utah (see “Subjects and Methods” section and table 1). DNA for 1 of the 467 patients was not available at the time the pools were constructed. This set of SNPs was highly enriched for missense, nonsense, and regulatory polymorphisms.16 The discovery scan on DNA pools yielded 1,654 markers with nominal P values <.05, 130 of which mapped within or near the MHC. These markers were then genotyped in pooled replication 1 samples consisting of 498 white, unrelated North American individuals with psoriasis and 498 white, unrelated North American controls matched for age and sex. Given that the MHC harbors the major susceptibility locus for psoriasis (the frequency of the psoriasis-risk HLA allele, C*0602, was 22.4% in our discovery cases vs. 8.5% in the controls; OR 3.10 [95% CI 2.37–4.08], P=2.37×10-17) and given the strong LD known to exist across this region,39 it is not surprising that 71 of the MHC-linked SNPs had P values <.05 in the replication 1 sample set, confirming the validity of this tiered approach to identify disease alleles. The most significant non–MHC-linked marker across both studies was rs3212227, an A→C SNP in the 3′ UTR of IL12B, located on 5q (pooled discovery samples P=5.24×10-5; pooled replication 1 samples P=1.88×10-8). This gene encodes the common IL-12p40 subunit of two cytokines, IL-12 and IL-23,14 and insertions and deletions in this gene have been associated with a rare, recessive form of Mendelian susceptibility to mycobacterial disease (MIM 209950).40,41
Individual genotyping of rs3212227 in the discovery and replication 1 samples and analyses of HWE showed that we could not reject HWE for this SNP in the cases and controls in either sample set (P=.847 in discovery cases and P=.588 in discovery controls; P=.297 in replication 1 cases and P=.913 in replication 1 controls) and confirmed association of the A (major) allele with risk of psoriasis (discovery sample set OR 1.59, P=1.89×10-4; replication 1 sample set OR 1.81, P=7.59×10-7), with a Fisher’s combined allelic P value of 3.39×10-9 and an ORcommon of 1.70 (table 2) (see below for a detailed discussion of experimentwide significance). The common A allele is increased in cases compared with controls (∼86% vs. ∼78%–79%, respectively). The association at rs3212227 appears to be independent of HLA-C, since stratification of the discovery data by presence or absence of the HLA-C*0602 risk allele did not result in significantly different ORs (1.47 and 1.61, respectively; Breslow-Day P=.88).
Table 2. .
Case-Control Allelic Association Analysis of rs3212227
| No. (Frequency) in |
||||
| Sample Set and Allele |
Cases | Controls | Risk Allele OR (95% CI)a |
Pb |
| Discoveryc: | ||||
| C | 133 (.143) | 192 (.209) | … | … |
| A | 799 (.857) | 726 (.791) | 1.59 (1.25–2.03) | 1.89×10−4 |
| Replication 1: | ||||
| C | 135 (.137) | 219 (.223) | … | … |
| A | 849 (.863) | 763 (.777) | 1.81 (1.42–2.28) | 7.59×10−7 |
| Combined | … | … | 1.70 (1.43–2.01) | 3.39×10−9 |
ORs for the discovery and replication 1 sample sets were determined for the common risk allele with the use of standard methods. For the combined analysis, we used the method of Mantel-Haenszel.
Allelic P values for the discovery and replication 1 sample sets were calculated using Fisher’s exact test. Allelic combined P values were determined using Fisher’s combined test.
Individual genotyping was performed for 467 cases and 460 controls from the discovery sample set.
Genotypic analysis of rs3212227 is presented in table 3. In a combined analysis of both sample sets, we found that the heterozygous genotype confers a modest, although not statistically significant, risk of psoriasis when compared with the minor homozygote genotype (ORcommon 1.47 [95% CI 0.86–2.50]), whereas individuals homozygous for the A allele appear to be at greater risk for psoriasis relative to the minor homozygote (ORcommon 2.55 [95% CI 1.52–4.28]). We also calculated Bayesian estimates of GRR (γ) for both the AA and CA genotypes (relative to the CC genotype) in each sample set, assuming that disease prevalence was beta distributed, with a mean of 2.3% (table 3). The resulting GRR estimates, which are quite similar to the ORs, are also consistent with a general model of inheritance, in which individuals carrying one copy of the A allele at rs3212227 have an increased risk of psoriasis relative to noncarriers, and individuals carrying two copies have an even greater risk.
Table 3. .
Case-Control Genotypic Association Analysis of rs3212227
| No. (Frequency) in |
||||
| Sample Set and Genotype(s) |
Cases | Controls | OR (95% CI)a | γb |
| Discoveryc,d: | ||||
| CC | 10 (.021) | 22 (.047) | … | … |
| CA | 113 (.242) | 148 (.322) | … | … |
| AA | 343 (.736) | 289 (.630) | … | … |
| CA vs. CC | … | … | 1.68 (.76–3.69) | 1.67 |
| AA vs. CC | … | … | 2.61 (1.22–5.60) | 2.57 |
| AA + CA vs. CC | … | … | 2.29 (1.07–4.90) | 2.27 |
| Replication 1e: | ||||
| CC | 12 (.024) | 24 (.049) | … | … |
| CA | 111 (.226) | 171 (.348) | … | … |
| AA | 369 (.750) | 296 (.603) | … | … |
| CA vs. CC | … | … | 1.30 (.62–2.70) | 1.29 |
| AA vs. CC | … | … | 2.49 (1.23–5.07) | 2.45 |
| AA + CA vs. CC | … | … | 2.06 (1.02–4.16) | 2.03 |
| Combinedf: | ||||
| CA vs. CC | … | … | 1.47 (.86–2.50) | … |
| AA vs. CC | … | … | 2.55 (1.52–4.28) | … |
| AA + CA vs. CC | … | … | 2.16 (1.29–3.63) | … |
ORs for the discovery and replication 1 sample sets were determined for the common risk allele with the use of standard methods. For the combined analysis, we used the method of Mantel-Haenszel.
GRR estimates for the indicated genotypes, calculated using a Bayesian approach as described in the “Subject and Methods” section.
Individual genotyping was performed for 467 cases and 460 controls from the discovery sample set.
P=.001, calculated using a Williams-corrected G test.
P=4.17×10-6, calculated using a Williams-corrected G test.
P=8.68×10-8, determined using Fisher's combined test.
Since false-positive results can be problematic in any large-scale experiment, we performed a Monte Carlo simulation, to obtain an experimentwide significance level. Two null hypotheses were considered, and results were presented under each. Under the first null model, which generates results similar to a Dunn-Sidak– or Bonferroni-corrected significance level,26 we assumed independence between the cases and controls, generating a uniform distribution of P values (see the “Association Statistics” subsection). The resulting combined P values, which represent 1,000 independent in silico experiments, ranged from 2.73×10-4 to 2.58×10-8 (uniform model) (fig. 1). None of these are as significant as the calculated Fisher’s combined P value of 3.39×10-9 for the IL12B 3′ UTR SNP, rs3212227. The second model (gamma model) (fig. 1), which uses the results from the pooled discovery samples to model the distribution of null P values and is more conservative,32 yielded 1,000 null combined P values ranging from 1.53×10-4 to 2.30×10-9. Only one exceeded the significance of the Fisher’s combined P value of 3.39×10-9 for the IL12B 3′ UTR SNP. Taken together, these data suggest that association of the IL12B 3′ UTR SNP with psoriasis is unlikely to be a type I error.
Figure 1. .

Results from the Monte Carlo simulation, showing the distribution of the 1,000 most significant combined P values under two null models. The arrow indicates the P value for rs3212227 (3.39×10-9).
IL12B Sequencing in Patients with Psoriasis
To identify novel IL12B SNPs in patients with psoriasis, we sequenced 1,136 bases of 5′ sequence, all exons and intron/exon boundaries, and 1,424 bases of 3′ sequence of IL12B in 96 individuals with psoriasis from the replication 1 sample set. On average, we successfully sequenced 89 individuals (coverage ranged from 70 to 96 individuals) and identified six SNPs, only one of which was not found in public databases. The minor allele of this novel 3′ UTR SNP (C158674941T; ss52085990) was observed on only one chromosome in the 96 individuals sequenced and was not genotyped further in this study.
Single-Marker Analysis of the IL12B Region
To further explore the association signal in the IL12B region, we first chose to determine whether other alleles within the IL12B region were associated with psoriasis by genotyping a combination of tag SNPs, which had the highest average power to detect disease-associated variants under a conservative disease model, and functional SNPs in our discovery and replication 1 sample sets. Of the 32 tag SNPs we selected using the program Redigo,21 from a total of 44 CEU phase I HapMap SNPs42 in a region of 364.8 kb surrounding IL12B on chromosome 5, 27 produced high-quality individual genotyping data in both the discovery and replication 1 sample sets, with minimal loss in average power (0.89 for 27 tag SNPs vs. 0.90 for 32 tag SNPs). We also individually genotyped a putative promoter polymorphism upstream of IL12B—rs17860508,43,44 which has been shown to affect IL-12 and IL-12Bp40 protein levels in stimulated cells, albeit inconsistently44–47—as well as two validated (SeattleSNPs resource) IL12B missense SNPs (rs3213119 and rs3213096) (fig. 2 and table 4), for a total of 30 additional SNPs in the IL12B region. An exact test of HWE of the genotypic data for these 30 variants, with the analysis performed separately for cases and controls in each sample set (data not shown), identified only two instances where a marker was not in HWE at the P<.05 significance level (rs7721001 in the discovery controls, P=.005; rs270654 in the replication cases, P=.020).
Figure 2. .

The physical map of the 360 kb around IL12B on human chromosome 5. Four known genes map to the area: EBF, FLJ31951, UBLCP1, and IL12B. AK097548 is a gene model with one mRNA as supporting evidence. Thirty-one markers spanning this region were genotyped in two independent case-control psoriasis sample sets. These markers included 27 tag SNPs, 1 putative promoter SNP (rs17860508), 2 missense SNPs (rs3213119 and rs3213096), and the original hit, rs3212227. On the basis of a visual examination of the CEU HapMap genotypes, 26 of the markers genotyped in the study map to a loosely defined block of LD. Blocks on either side of this central region were queried using five additional markers.
Table 4. .
MAFs and Allele-Based Association of IL12B-Associated SNPs with Psoriasis[Note]
| Discovery Sample Seta |
Replication 1 Sample Set |
Replication 2 Sample Set |
Combined Analysis |
||||||||||||||
| Frequency in |
Frequency in |
Frequency in |
|||||||||||||||
|
dbSNP Identification Number |
Gene | Type | Position and Allelesb |
Cases (n=467) | Controls (n=460) | OR | Allelic Pc | Cases (n=498) | Controls (n=498) | OR | Allelic Pc | Cases (n=481) | Controls (n=424) | OR | Allelic Pc | ORcommon (95% CI)d | Pcombe |
| rs929779 | EBF | Intron | T158420776G | .465 | .452 | 1.05 | .608 | .488 | .487 | 1.00 | 1.000 | … | … | … | … | … | … |
| rs1422668 | EBF | Intron | G158438712A | .310 | .324 | .94 | .549 | .343 | .337 | 1.03 | .813 | … | … | … | … | … | … |
| rs6898290 | EBF | Intron | C158446124T | .415 | .413 | 1.01 | .925 | .453 | .441 | 1.05 | .619 | … | … | … | … | … | … |
| rs2161357 | … | Intergenic | C158469697T | .193 | .215 | .88 | .273 | .218 | .198 | 1.13 | .293 | … | … | … | … | … | … |
| rs6897374 | … | Intergenic | A158473778C | .090 | .074 | 1.24 | .236 | .084 | .103 | .80 | .165 | … | … | … | … | … | … |
| rs6896438 | … | Intergenic | C158480454G | .390 | .400 | .96 | .669 | .398 | .391 | 1.03 | .748 | … | … | … | … | … | … |
| rs270661 | … | Intergenic | A158492732G | .197 | .200 | .98 | .907 | .233 | .183 | 1.36 | .007 | … | … | … | … | … | … |
| rs270654 | … | Intergenic | G158497687A | .109 | .103 | 1.07 | .651 | .134 | .110 | 1.27 | .100 | … | … | … | … | … | … |
| rs6556398 | … | Intergenic | A158505815C | .321 | .304 | 1.08 | .452 | .297 | .320 | .90 | .285 | … | … | … | … | … | … |
| rs717925 | … | Intergenic | C158513109G | .014 | .015 | .91 | .848 | .010 | .028 | .35 | .005 | … | … | … | … | … | … |
| rs10035989 | FLJ31951 | UTR | C158517569T | .198 | .190 | 1.06 | .638 | .194 | .185 | 1.06 | .607 | … | … | … | … | … | … |
| rs2043270 | FLJ31951 | Intron | G158530559A | .088 | .075 | 1.18 | .350 | .081 | .071 | 1.15 | .448 | … | … | … | … | … | … |
| rs1897565 | FLJ31951 | Intron | C158550843T | .212 | .273 | .71 | .002 | .243 | .280 | .83 | .066 | … | … | … | … | … | … |
| rs7721001 | … | Intergenic | T158583392C | .418 | .466 | .82 | .039 | .477 | .470 | 1.03 | .787 | … | … | … | … | … | … |
| rs11744690 | … | Intergenic | C158619731T | .345 | .329 | 1.07 | .489 | .323 | .307 | 1.08 | .440 | … | … | … | … | … | … |
| rs1368437 | MGC10067 | Intron | G158639557C | .118 | .094 | 1.29 | .097 | .130 | .101 | 1.33 | .049 | … | … | … | … | … | … |
| rs3212227 | IL12B | UTR | C158675528A | .143 | .209 | .63 | 1.89×10−4 | .137 | .223 | .55 | 7.59×10−7 | .167 | .212 | .74 | .014 | .64 (.56–.73) | 7.85×10−10 |
| rs3213119 | IL12B | F298V | T158676366G | .031 | .027 | 1.15 | .680 | .030 | .039 | .77 | .327 | … | … | … | … | … | … |
| rs3213096 | IL12B | I33V | A158682907G | .000 | .003 | NC | .122 | .002 | .000 | NC | .248 | … | … | … | … | … | … |
| rs3212220 | IL12B | Intron | T158686773G | .144 | .207 | .64 | 3.81×10−4 | .141 | .225 | .56 | 1.37×10−6 | … | … | … | … | … | … |
| rs1433048 | IL12B | Intron | G158688423A | .213 | .185 | 1.19 | .146 | .212 | .190 | 1.15 | .239 | … | … | … | … | … | … |
| rs17860508f | IL12B | Promoter | GC158692778TTAGAG | .453 | .498 | .83 | .056 | .462 | .511 | .82 | .031 | … | … | … | … | … | … |
| rs7709212g | … | Intergenic | C158696755T | .272 | .333 | .75 | .005 | .267 | .351 | .68 | 6.65×10−5 | … | … | … | … | … | … |
| rs953861g | … | Intergenic | G158705160A | .190 | .169 | 1.15 | .250 | .220 | .179 | 1.30 | .025 | … | … | … | … | … | … |
| rs6869411g | … | Intergenic | C158714182T | .390 | .388 | 1.01 | .962 | .358 | .339 | 1.09 | .421 | … | … | … | … | … | … |
| rs1833754 | … | Intergenic | G158751505A | .042 | .052 | .79 | .323 | .047 | .060 | .78 | .198 | … | … | … | … | … | … |
| rs6887695 | … | Intergenic | C158755223G | .254 | .319 | .73 | .002 | .242 | .333 | .64 | 9.77×10−6 | .251 | .308 | .75 | .007 | .70 (.63–.79) | 4.08×10−8 |
| rs918520 | … | Intergenic | C158758888G | .253 | .203 | 1.33 | .011 | .246 | .216 | 1.18 | .123 | … | … | … | … | … | … |
| rs4921226 | … | Intergenic | A158763397G | .317 | .340 | .90 | .320 | .365 | .328 | 1.18 | .094 | … | … | … | … | … | … |
| rs1422878 | … | Intergenic | A158771795G | .355 | .369 | .94 | .562 | .296 | .376 | .70 | 1.73×10−4 | … | … | … | … | … | … |
| rs4921496 | … | Intergenic | T158780649C | .274 | .249 | 1.14 | .245 | .291 | .265 | 1.14 | .193 | … | … | … | … | … | … |
Note.— The data used to calculate frequencies are from individual genotyping. Results are reported for the minor allele. NC = not calculated.
Individual genotyping was performed for 467 cases and 460 controls from the discovery sample set.
Positions according to genomic contig NT_023133 (Entrez Nucleotide). The minor allele is listed first, followed by the position in National Center for Biotechnology Information Genome Build 35.1 and then the major allele.
Calculated using Fisher’s exact test.
Calculated using a Mantel-Haenszel common OR.
Calculated using Fisher’s combined test.
Putative promoter polymorphism.43
SNP is located within AK097548, a gene model with one mRNA as supporting evidence.
All 30 variants were analyzed for single-marker association with psoriasis susceptibility. In addition to the original hit, three other SNPs—rs3212220, rs7709212, and rs6887695, all of which lie upstream of the IL12B ATG start codon—had P values <.05 in an allelic test, with consistent ORs (i.e., the psoriasis-risk allele was the same) in both sample sets (table 4). The results for the putative promoter polymorphism, rs17860508, did not reach statistical significance in the discovery sample set (P=.056) and were only marginally significant in the replication 1 sample set (P=.031). The two missense SNPs, which were relatively infrequent in our sample sets (rs3213119, F298V: allele frequency ∼3%–4%; rs3213096, I33V: allele frequency <1%), were not significantly associated with disease.
LD and Haplotype Analysis of the IL12B-Region SNPs
We examined the LD patterns in this region by calculating the pairwise r2 values between all 31 markers separately for the cases and the controls in each study (fig. 3). The original hit, rs3212227, is in high LD with one of the tag SNPs, rs3212220 (r2>0.97 in the cases and the controls of both sample sets), and is in modest LD with rs1897565 (r2=0.25–0.33), rs17860508 (r2=0.12–0.23), rs7709212 (r2=0.28–0.40), and rs6887695 (r2=0.15–0.31) but exhibits little to no LD with the other 25 SNPs. Four other marker pairs show moderate LD (0.50<r2<0.80) in the cases and the controls of both sample sets; however, the majority of the markers exhibited minimal LD between one another.
Figure 3. .

Pairwise LD between the 31 IL12B-region SNPs, as measured by r2 in the cases and controls of both the discovery and replication 1 sample sets. The indexes of the SNPs (table 4) were arranged vertically from SNP 2 to SNP 31 and horizontally from SNP 1 to SNP 30.
To focus on regions of interest, we generated three-marker sliding-window haplotypes for the 28 markers with a MAF >5% for the cases and the controls from both sample sets. Global P values were determined using the Haplo.Stats program,24 to assess the overall haplotype-frequency differences between the cases and the controls for each 3-SNP window (fig. 4). Two peaks with highly significant global P values (P<.0001) were observed in both sample sets. The first peak (w14-17) centers on the original marker, rs3212227, and on rs3212220, which, as noted above, are in strong LD with one another. The second peak (w22-24) centers on rs6887695. These three markers also showed the most-significant single-marker association (table 4).
Figure 4. .

3-SNP sliding-window analyses of the IL12B-region SNPs
On the basis of these results, we focused subsequent analyses on the 13 SNPs with a MAF >5% and encompassed by these two peaks (rs11744690 to rs4921226 in table 4). Haplotypes for all 13 SNPs across this 144-kb region were predicted, and association between these haplotypes and disease status was determined using Haplo.Stats. This was followed by the stepwise removal of each SNP and reanalysis of the haplotypes and their association with disease, to define the minimum number of SNPs that explained the observed association (data not shown). This systematic approach indicated that 2 of these 13 SNPs—our original hit (rs3212227) and rs6887695, which exhibit some LD (r2=0.15–0.31)—could explain the majority, if not all, of the association with psoriasis observed in both sample sets (table 5). The 2-SNP haplotype containing the major allele of both SNPs (A at rs3212227 and G at rs6887695) appears to be associated with risk in both sample sets (discovery sample set OR 1.42, P=3.27×10-4; replication 1 sample set OR 1.48, P=1.35×10-5), whereas a second haplotype containing the minor allele at both SNPs (C-C) confers protection (discovery sample set OR 0.63, P=5.8×10-4; replication 1 sample set OR 0.46, P=2.56×10-9). The frequencies of the other two haplotypes, which contain a risk allele at one SNP and a protective allele at the second SNP, did not exhibit substantial differences between cases and controls.
Table 5. .
Two-Marker Haplotypes for the IL12B Region[Note]
| Discovery Sample SetGlobal P=.001 |
Replication 1 Sample SetGlobal P=5.13×10-8 |
Replication 2 Sample SetGlobal P=.029 |
Combined AnalysisGlobala Pcomb=5.94×10-10 |
||||||||||||
| Haplotype |
No. (Frequency) in |
No. (Frequency) in |
No. (Frequency) in |
||||||||||||
| rs3212227 | rs6887695 | Case (n=467) | Control (n=460) | OR | P | Case (n=498) | Control (n=498) | OR | P | Case (n=481) | Control (n=424) | OR | P | ORcommon (95%CI)b |
Pcomba |
| A | G | 667 (.714) | 587 (.638) | 1.42 | 3.27×10−4 | 716 (.719) | 631 (.634) | 1.48 | 1.35×10−5 | 678 (.705) | 548 (.646) | 1.31 | 6.00×10−3 | 1.40 (1.25–1.57) | 8.11×10−9 |
| A | C | 134 (.144) | 141 (.153) | .93 | .454 | 144 (.145) | 143 (.142) | 1.01 | .874 | 124 (.129) | 120 (.142) | .90 | .345 | .95 (.82–1.10) | .680 |
| C | C | 103 (.110) | 152 (.166) | .63 | 5.80×10−4 | 96 (.096) | 188 (.190) | .46 | 2.56×10−9 | 117 (.122) | 140 (.165) | .70 | 7.00×10−3 | .58 (.50–.68) | 5.65×10−12 |
| C | G | 30 (.032) | 40 (.043) | .73 | .145 | 40 (.040) | 34 (.035) | 1.18 | .731 | 43 (.045) | 40 (.047) | .95 | .693 | .94 (0.72–1.24) | .516 |
Note.— A pseudo-Gibbs sampling algorithm from the program SNPAnalyzer was used to estimate haplotype frequencies from unphased data, with cases and controls treated separately. The Haplo.Stats package was used to test for association between haplotypes and disease status.
Calculated using Fisher’s combined test.
Calculated using a Mantel-Haenszel common OR.
Because these two markers (rs3212227 and rs6887695) exhibited some LD, we performed genotype-conditioned analyses similar to that described by Thomson and colleagues,35,36 to see if either SNP was associated with disease independent of the other SNP. To do this, we calculated a summary statistic for each sample set, using the genotypes at one marker conditioned on the other marker, and evaluated the significance of these observed results through a permutation test. The results of 20,000 permutations for each sample set demonstrated that association of rs3212227 with psoriasis given rs6887695 (and rs6887695 given rs3212227) was significant at the .001 level in both sample sets, indicating that each SNP contributes independently to psoriasis risk.
Replication of IL12B Results in a Third Sample Set and in Combined Analyses
We then genotyped rs3212227 and rs6887695 in a third independent sample set consisting of 481 white North American individuals with psoriasis and 424 white North American controls. Allele frequencies were comparable across all three sample sets, and single-marker analysis confirmed that both rs3212227 and rs6887695 were associated with psoriasis in this independent sample set (P=.014 and P=.007, respectively) (table 4). The combined analysis for each individual SNP across all three sample sets was highly significant (combined P [Pcomb]=7.85×10-10 and Pcomb=4.08×10-8, respectively). Haplotypes were also estimated in the third sample set, confirming the common IL12B risk haplotype (OR 1.31, P=6.00×10-3) as well as the less frequent protective haplotype (OR 0.70, P=7.00×10-3) (table 5). The combined analyses across all three sample sets were highly significant; the frequency of the common risk A-G haplotype is ∼71%–72% in cases versus ∼64% in controls (ORcommon 1.40, Pcomb=8.11×10-9), and the frequency of the protective C-C haplotype is ∼10%–12% in cases versus ∼17%–19% in controls (ORcommon 0.58, Pcomb=5.65×10-12) (table 5). Importantly, the other two haplotypes carrying a risk allele at one SNP and a protective allele at the other SNP were not associated with psoriasis. Together, these data provide convincing statistical evidence that the IL12B region on 5q31.1-33.1 harbors a psoriasis-susceptibility locus.
Analysis of IL12B-Related Genes
IL12B encodes the common IL-12p40 subunit of two heterodimeric cytokines, IL-12 and IL-23, each with a distinct subunit encoded by the genes IL12A (IL-12p35) and IL23A (IL-23p19). The IL-12 and IL-23 receptors also share a common subunit encoded by IL12RB1, in addition to their unique components—IL23R for the IL-23 receptor and IL12RB2 for the IL-12 receptor. To determine whether variants of these candidate genes might be associated with psoriasis, we individually genotyped 17 SNPs (7 in IL12A, 2 in IL23A, 4 in IL12RB1, 1 in IL12RB2, and 3 in IL23R) from our collection of assays in the discovery sample set. Two SNPs, rs7530511 in IL23R (OR 0.67, P=.006) and rs2914119 in IL12A (OR 0.78, P=.045), were nominally significant in the single-marker analyses (P<.05) (table 6). We also estimated haplotypes for all SNPs in each gene (data not shown) and tested for association with disease with the use of Haplo.Stats. Since IL23R and IL12RB2 lie directly adjacent to each another on chromosome 1, all four SNPs in these two genes were used together in the haplotype analysis of this region. If the global P value for all the SNPs in a particular gene was significant, we then evaluated the contribution of each SNP to this signal, using a systematic stepwise approach in which each SNP was removed and Haplo.Stats was rerun. Using this approach, we found that a second IL23R SNP, rs11209026, in combination with rs7530511, was associated with psoriasis risk (global P=.004) (table 7). The most common haplotype marked by these two SNPs, C at rs7530511 and G at rs11209026, was increased in patients compared with controls (85.2% vs. 79.3%, OR 1.5, P=9.48×10-4) (table 7) and was more significant than each SNP individually (table 6).
Table 6. .
Allele Frequencies and Allele-Based Association of Markers in IL12B-Related Genes
| Discovery Sample Set |
Replication 1 Sample Set |
Replication 2 Sample Set |
Combined Analysis |
|||||||||||||||
| Frequency in |
Frequency in |
Frequency in |
||||||||||||||||
| dbSNP Identification Number |
Locus | Type | Chromosome | Positiona | Cases (n=467) | Controls (n=460) | OR | Allelic Pb | Cases (n=498) | Controls (n=498) | OR | Allelic Pb | Cases (n=481) | Controls (n=424) | OR | Allelic Pb | ORcommon (95%CI)c | Pcombd |
| rs1884444 | IL23R | H3Q | 1 | G67345833T | .451 | .476 | .90 | .284 | … | … | … | … | … | … | … | … | … | … |
| rs7530511 | IL23R | L310P | 1 | T67397408C | .103 | .146 | .67 | .006 | .100 | .114 | .87 | .347 | .108 | .130 | .81 | .166 | .78(.66–.91) | .014 |
| rs11209026 | IL23R | Q381R | 1 | A67417979G | .044 | .060 | .73 | .142 | .051 | .077 | .65 | .004 | .035 | .066 | .52 | .003 | .63(.50–.79) | 1.89×10−4 |
| rs1109918 | IL12RB2 | 3′ UTR | 1 | C67574503T | .026 | .024 | 1.08 | .882 | … | … | … | … | … | … | … | … | … | … |
| rs583911 | IL12A | Intron | 3 | G161193092A | .421 | .449 | .89 | .241 | … | … | … | … | … | … | … | … | … | … |
| rs2227314 | IL12A | Intron | 3 | T161194756G | .422 | .448 | .89 | .259 | … | … | … | … | … | … | … | … | … | … |
| rs2243131 | IL12A | Intron | 3 | C161194760A | .161 | .139 | 1.18 | .216 | … | … | … | … | … | … | … | … | … | … |
| rs2243149 | IL12A | Intergenic | 3 | T161198414C | .419 | .413 | 1.02 | .814 | … | … | … | … | … | … | … | … | … | … |
| rs2243154 | IL12A | Intergenic | 3 | A161198944G | .086 | .080 | 1.07 | .737 | … | … | … | … | … | … | … | … | … | … |
| rs6771983 | IL12A | Intergenic | 3 | T161221155C | .007 | .004 | 1.73 | .548 | … | … | … | … | … | … | … | … | … | … |
| rs2914119 | IL12A | Intergenic | 3 | T161227140C | .160 | .196 | .78 | .045 | .191 | .190 | 1.01 | .955 | … | … | … | … | … | … |
| rs2371494 | IL23A | Intergenic | 12 | A55014267G | .058 | .074 | .77 | .161 | … | … | … | … | … | … | … | … | … | … |
| rs11171806 | IL23A | S106S | 12 | A55019798G | .054 | .073 | .72 | .104 | … | … | … | … | … | … | … | … | … | … |
| rs401502 | IL12RB1 | G378R | 19 | G18041413C | .300 | .313 | .94 | .579 | … | … | … | … | … | … | … | … | … | … |
| rs375947 | IL12RB1 | T365M | 19 | G18041451A | .301 | .313 | .95 | .614 | … | … | … | … | … | … | … | … | … | … |
| rs11575926 | IL12RB1 | H156R | 19 | A18049408G | .176 | .174 | 1.01 | .951 | … | … | … | … | … | … | … | … | … | … |
| rs393548 | IL12RB1 | Intergenic | 19 | A18058744T | .206 | .210 | .97 | .819 | … | … | … | … | … | … | … | … | … | … |
The minor allele is listed first, followed by the position in Genome Build 35.1 and then the major allele.
Calculated using Fisher’s exact test.
Calculated using a Mantel-Haenszel common OR.
Calculated using Fisher’s combined test.
Table 7. .
Two-Marker Haplotypes for the IL23R Gene[Note]
| Discovery Sample SetGlobal P=.004 |
Replication 1 Sample SetGlobal P=.002 |
Replication 2 Sample SetGlobal P=.003 |
Combined AnalysisGlobala Pcomb=4.14×10-6 |
||||||||||||
| Haplotype |
No. (Frequency) in |
No. (Frequency) in |
No. (Frequency) in |
||||||||||||
| rs7530511 | rs11209026 | Case (n=467) | Control (n=460) | OR | P | Case (n=498) |
Control (n=498) | OR | P | Case (n=481) | Control (n=424) | OR | P | ORcommon (95%CI)b | Pcomba |
| C | G | 796 (.852) | 730 (.793) | 1.50 | 9.48×10−4 | 840 (.843) | 794 (.797) | 1.37 | .006 | 824 (.857) | 682 (.804) | 1.45 | .003 | 1.44 (1.25–1.65) | 3.13×10−6 |
| T | G | 96 (.103) | 135 (.147) | .67 | .005 | 98 (.098) | 113 (.113) | .85 | .249 | 104 (.108) | 110 (.130) | .81 | .003 | .77 (.65–.91) | 3.22×10−4 |
| C | A | 42 (.045) | 55 (.060) | .74 | .134 | 56 (.056) | 89 (.089) | .61 | .004 | 34 (.035) | 56 (.066) | .52 | .153 | .62 (.49–.77) | .005 |
| T | A | 0 | 0 | … | … | 2 (.002) | 0 | … | … | 0 | 0 | … | … | … | … |
Note.— A pseudo-Gibbs sampling algorithm from the program SNPAnalyzer was used to estimate haplotype frequencies from unphased genotyping data, with cases and controls treated separately. The Haplo.Stats package was used to test for association between haplotypes and disease status.
Calculated using a Mantel-Haenszel common OR.
Calculated using Fisher’s combined test.
The two IL23R SNPs and the IL12A SNP were then individually genotyped in the replication 1 sample set (table 6). The IL12A SNP did not replicate (P=.955), and a meta-analysis of both discovery and replication 1 samples yielded a Fisher’s combined P value of .178 (data not shown). However, the common IL23R haplotype, C at rs7530511 and G at rs11209026, was associated with psoriasis risk in the replication 1 sample set (OR 1.37, P=.006), as well as in the replication 2 sample set (OR 1.45, P=.003) (table 7), and the combined analysis across all three sample sets was highly significant (ORcommon 1.44, Pcomb=3.13×10-6), providing strong statistical evidence for a psoriasis-susceptibility gene on 1p31. It is interesting to note that these two IL23R SNPs are both missense SNPs (rs7530511, L310P; rs11209026, Q381R) with an r2 value between them of <0.012 in the cases and controls of all three sample sets (data not shown). The common risk haplotype carries a proline at amino acid 310 and an arginine at amino acid 381. Both of these amino acids are conserved in the chimpanzee, mouse, rat, cow, dog, and chicken.
Additional Statistical Support of IL12B and IL23R Haplotypes
Complementing traditional homogeneity tests, significant differences in LD patterns between cases and controls can also indicate disease association. We applied the LD contrast test described by Nielsen and colleagues28 to pairs of SNPs in IL12B and IL23R. The results show departures between case and control pairwise values of D at rs3212227-rs6887695 for IL12B, as expected under a disease model: Pcs-dis,ct-dis=.0129, Pcs-rep1,ct-rep1=5.16×10-8, Pcs-rep2,ct-rep2=.0497 (where “cs” denotes cases,“ct” denotes controls, “dis” denotes the discovery sample, and “rep1” and “rep2” denote replication sample sets 1 and 2, respectively), and Pcomb=1.05×10-8. With regard to rs7530511-rs11209026 at IL23R, this test yielded similar significant P values: Pcs-dis,ct-dis=.0085, Pcs-rep1,ct-rep1=.0019, Pcs-rep2,ct-rep2=.0019, and Pcomb=5.30×10-6. Since population stratification and other sampling irregularities can generate departures of pairwise LD measures from their null distribution, we performed the LD contrast test on all combinations of control-control (i.e., discovery controls vs. replication 1 controls, discovery controls vs. replication 2 controls, etc.), case-case, and case-control samples, to better understand the significance of our results. In all instances for both genes, case-control comparisons were statistically significant (P<.05), whereas the case-case and control-control comparisons were not (results not shown).
Bayes factor curves were calculated for the risk rs3212227-rs6887695 IL12B haplotype and the risk rs7530511-rs11209026 IL23R haplotype. Bayes factors were adjusted for population stratification and were combined across the three independent studies.33 The results show a maximum log10B(R) of 6.85 for the IL12B haplotype data, occurring at R=1.41. Given equal prior probabilities for disease and null models, the probability of the disease hypothesis, given the data, is ∼7-million-fold larger (at a disease model of R=1.41) than the probability of the null hypothesis conditioned on the data. Further, the log10B(R)>4, as R spans 1.15 to 1.73, testifying to the strength, robustness, and specificity of the result. Similar results were observed for the IL23R haplotype data, with a maximum log10B(R) of 5.78 at R=1.47. Log10B(R) remains >4 for the relatively narrow range of 1.20<R<1.80.
Diplotype Analyses
To better understand how the IL12B and IL23R genes influence an individual’s risk of psoriasis, we assessed the impact of diplotypes at each locus independently and then jointly on psoriasis risk. Of the 10 possible 2-SNP IL12B diplotypes, 9 were observed in our samples (table 8). The combined results from all three sample sets suggest that there is a hierarchy of risk depending on an individual’s IL12B diplotype. Psoriasis susceptibility appears to be conferred by two copies of the common risk haplotype (A-G/A-G ORcommon 1.52, Pcomb=6.14×10-7), which is found in ∼48%–51% of cases versus 38%–40% of controls. One copy of the risk haplotype (A-G) in combination with a copy of a “neutral” haplotype (A-C or C-G) does not appear to substantially affect psoriasis risk; however, individuals who carry copies of both the risk and the protective haplotypes (A-G/C-C) appear to be protected (ORcommon 0.67, Pcomb=1.39×10-4). Individuals homozygous for the protective haplotype (C-C/C-C) are relatively uncommon (∼1% of cases vs. ∼2–3% of controls); however, our data suggest that their risk of psoriasis may be even lower (ORcommon 0.38, Pcomb=.018), although, given the stochastic variability around this estimate, additional samples are required to fully address this issue. The population-attributable fraction30,31 summarized across all three sample sets for the homozygous susceptible A-G/A-G IL12B diplotype, relative to all other diplotypes collapsed together, was calculated to be 17.1% (95% CI 11.4%–22.5%).
Table 8. .
Diplotype Analysis for the IL12B SNPs rs3212227 and rs6887695
| Discovery Sample SetGlobala P=.033 |
Replication 1 Sample SetGlobala P=3.21×10-7 |
Replication 2 Sample SetGlobala P=.042 |
Combined AnalysisGlobalb P=1.13×10-7 |
|||||||||||
| No. (%) of |
No. (%) of |
No. (%) of |
||||||||||||
| Diplotypec | Cases | Controls | OR | Pd | Cases | Controls | OR | Pd | Cases | Controls | OR | Pd | ORcommon (95%CI)e | Pcombf |
| A-G/A-G | 238 (.510) | 186 (.403) | 1.53 | 1.54×10−3 | 252 (.506) | 190 (.382) | 1.66 | 9.81×10−5 | 232 (.482) | 171 (.403) | 1.38 | .019 | 1.52 (1.31–1.77) | 6.14×10−7 |
| A-G/C-C | 75 (.161) | 96 (.209) | .73 | .063 | 73 (.147) | 121 (.243) | .54 | 1.60×10−4 | 84 (.175) | 91 (.215) | .77 | .130 | .67 (.55–.81) | 1.39×10−4 |
| A-G/A-C | 91 (.195) | 90 (.196) | .99 | 1 | 107 (.215) | 104 (.209) | 1.04 | .877 | 99 (.206) | 81 (.191) | 1.10 | .617 | 1.04 (.87–1.25) | … |
| A-G/C-G | 25 (.054) | 29 (.063) | .84 | .576 | 32 (.064) | 26 (.052) | 1.25 | .499 | 31 (.064) | 34 (.080) | .79 | .370 | .94 (.69–1.27) | … |
| C-C/C-C | 6 (.013) | 13 (.028) | .45 | .109 | 4 (.008) | 16 (.032) | .24 | .011 | 5 (.010) | 8 (.019) | .55 | .402 | .38 (.19–.68) | .018 |
| C-C/A-C | 13 (.028) | 23 (.050) | .54 | .090 | 7 (.014) | 27 (.054) | .25 | 6.70×10−4 | 13 (.027) | 27 (.064) | .41 | .009 | .39 (.25–.59) | 6.51×10−5 |
| C-C/C-G | 3 (.006) | 7 (.015) | .42 | .221 | 8 (.016) | 8 (.016) | 1.00 | 1 | 10 (.021) | 6 (.014) | 1.48 | .615 | .95 (.50–1.80) | … |
| A-C/A-C | 15 (.032) | 14 (.031) | 1.06 | 1 | 15 (.030) | 6 (.012) | 2.55 | .075 | 6 (.012) | 6 (.014) | .88 | 1 | 1.35 (.81–2.33) | .522 |
| A-C/C-G | 0 | 0 | … | … | 0 | 0 | … | … | 0 | 0 | … | … | … | … |
| C-G/C-G | 1 (.002) | 2 (.004) | .49 | .622 | 0 | 0 | … | … | 1 (.002) | 0 | … | 1 | … | … |
Calculated using a Williams-corrected G test.
Calculated using Fisher’s combined test.
Allele 1 rs3212227-allele 1 rs6887695/allele 2 rs3212227-allele 2 rs6887695.
Calculated using Fisher’s exact test.
Calculated using a Mantel-Haenszel common OR.
Calculated for diplotypes with the same effect (risk or protection) in all three sample sets, with use of Fisher’s combined test.
At the IL23R locus, 7 of the 10 possible 2-SNP diplotypes were observed (table 9). The data from all three sample sets suggest that individuals homozygous for the susceptible C-G haplotype are at increased risk of psoriasis (ORcommon 1.48, Pcomb=2.24×10-5), whereas all other diplotypes appear to be neutral or protective. The estimated population-attributable fraction summarized across all three sample sets for the IL23R homozygous risk diplotype (C-G/C-G), relative to all other diplotypes collapsed together, is 23.5% (95% CI 14.9%–31.5%).
Table 9. .
Diplotype Analysis for the IL23R SNPs rs7530511 and rs11209026
| Discovery Sample SetGlobala P=.033 |
Replication 1 Sample SetGlobala P=8.47×10-6 |
Replication 2 Sample SetGlobala P=.009 |
Combined AnalysisGlobalb P=5.45×-7 |
|||||||||||
| No. (%) of |
No. (%) of |
No. (%) of |
||||||||||||
| Diplotypec | Cases | Controls | OR | Pd | Cases | Controls | OR | Pd | Cases | Controls | OR | Pd | ORcommon(95% CI)e | Pcombf |
| C-G/C-G | 340 (.728) | 288 (.626) | 1.60 | 9.58×10−4 | 357 (.717) | 316 (.635) | 1.46 | 6.73×10−3 | 350 (.728) | 279 (.658) | 1.39 | .025 | 1.48 (1.26–1.74) | 2.24×10−5 |
| C-G/T-G | 84 (.180) | 112 (.243) | .68 | .020 | 81 (.163) | 90 (.181) | .88 | .502 | 94 (.195) | 81 (.191) | 1.03 | .933 | .85 (.70–1.02) | … |
| C-G/C-A | 33 (.071) | 43 (.093) | .74 | .232 | 45 (.090) | 72 (.145) | .59 | .010 | 30 (.062) | 43 (.101) | .59 | .037 | .63 (.49–.82) | .005 |
| C-G/T-A | 0 | 0 | … | … | 0 | 0 | … | … | 0 | 0 | … | … | … | … |
| C-A/C-A | 2 (.004) | 2 (.004) | .98 | 1 | 1 (.002) | 5 (.010) | .20 | .217 | 1 (.002) | 2 (.005) | .44 | .602 | .43 (.08–1.38) | .667 |
| C-A/T-G | 4 (.009) | 8 (.017) | .47 | .260 | 7 (.014) | 7 (.014) | 1.00 | 1.000 | 2 (.004) | 9 (.021) | .19 | .029 | .52 (.24–1.00) | .136 |
| C-A/T-A | 0 | 0 | … | … | 2 (.004) | 0 | … | .499 | 0 | 0 | … | … | … | … |
| T-G/T-G | 4 (.009) | 7 (.015) | .56 | .382 | 5 (.010) | 8 (.016) | .62 | .579 | 4 (.008) | 10 (.024) | .35 | .102 | .49 (.23–.95) | .270 |
| T-G/T-A | 0 | 0 | … | … | 0 | 0 | … | … | 0 | 0 | … | … | … | … |
| T-A/T-A | 0 | 0 | … | … | 0 | 0 | … | … | 0 | 0 | … | … | … | … |
Because of small counts in some of the cells, as well as the distribution of these counts, global P values were obtained by performing a permutation procedure on the data and generating a log-likelihood ratio homogeneity statistic for each permutation. The convergence of the statistic to its limiting distribution was measured by plotting the first two central moments as a function of the number of replicates. When the number of replicates reached a number in which the error of the P value estimates from a subsequent modeling procedure was negligible (<1%), a gamma distribution was fit using maximum-likelihood estimates for the parameters. Integration of the resulting gamma distribution yielded the global P values.
Calculated using Fisher’s combined test.
Allele 1 rs7530511-allele 1 rs11209026/allele 2 rs7530511-allele 2 rs11209026.
Calculated using Fisher’s exact test.
Calculated using a Mantel-Haenszel common OR.
Calculated for diplotypes with the same effect (risk or protection) in all three sample sets, with use of Fisher’s combined test.
To assess the joint impact of the IL12B and IL23R susceptible haplotypes, we determined the distribution of diplotypes at both genes, comparing cases and controls in all three sample sets (table 10). Given the large number of possible diplotypes across these two loci relative to the number of cases and controls, we chose to concentrate our analysis on the risk haplotypes at each locus (A-G for IL12B and C-G for IL23R) and to group the three nonrisk haplotypes at each locus into a single haplotype (indicated as “X” for both loci in table 10). The combined analysis shows that individuals homozygous for both the IL12B and IL23R predisposing haplotypes, ∼35%–37% of cases versus ∼24%–27% of controls, appear to be at increased risk of psoriasis (ORcommon 1.66, Pcomb=1.33×10-8). A much larger sample size is needed to thoroughly evaluate the effect of all possible two-locus diplotypes; however, a preliminary meta-analysis of the available data (using a Bayesian approach, assuming no epistasis between loci and an expected prevalence of psoriasis equal to 2% in the general population) suggests at least a threefold difference in risk between individuals who carry one copy of the protective C-C IL12B haplotype and one copy of the protective C-A IL23R haplotype, representing 4.4% of the general population (C-C/X–C-A/X, expected psoriasis risk 0.84%) relative to individuals who are homozygous for risk haplotypes at both loci, representing 25.5% of the general population (A-G/A-G–C-G/C-G, expected psoriasis risk 2.83%).
Table 10. .
Two-Locus Diplotypes for IL12B and IL23R
| Discovery Sample SetGlobala P=6.16×10-5 |
Replication 1 Sample SetGlobala P=9.72×10-4 |
Replication 2 Sample SetGlobala P=.005 |
Combined AnalysisGlobalb P=7.88×10-8 |
|||||||||||
| No. (%) of |
No. (%) of |
No. (%) of |
||||||||||||
| Two-Locus Diplotypec |
Cases | Controls | OR | Pd | Cases | Controls | OR | Pd | Cases | Controls | OR | Pd | ORcommon (95% CI)e | Pcombf |
| A-G/A-G–C-G/C-G | 175 (.375) | 110 (.239) | 1.91 | 9.62×10−6 | 174 (.350) | 121 (.243) | 1.67 | 2.98×10−4 | 168 (.349) | 116 (.274) | 1.43 | 1.50×10−2 | 1.66 (1.41–1.95) | 1.33×10−8 |
| A-G/A-G–C-G/X | 56 (.120) | 69 (.150) | .77 | .211 | 68 (.137) | 62 (.125) | 1.11 | .638 | 59 (.123) | 48 (.113) | 1.10 | .681 | … | … |
| A-G/A-G–X/X | 7 (.015) | 7 (.015) | .98 | 1 | 9 (.018) | 7 (.014) | 1.29 | .802 | 4 (.008) | 7 (.017) | .50 | .364 | … | … |
| A-G/X–C-G/C-G | 139 (.298) | 145 (.315) | .92 | .569 | 159 (.320) | 160 (.322) | .99 | 1 | 154 (.320) | 138 (.325) | .98 | .887 | .96 (.82–1.13) | .968 |
| A-G/X–C-G/X | 51 (.109) | 61 (.133) | .80 | .314 | 48 (.097) | 80 (.161) | .56 | .003 | 57 (.119) | 56 (.132) | .88 | .547 | .73 (.58–.91) | .019 |
| A-G/X–X/X | 1 (.002) | 9 (.020) | .11 | .011 | 5 (.010) | 10 (.020) | .49 | .298 | 3 (.006) | 12 (.028) | .22 | .016 | .27 (.11–.53) | .003 |
| X/X–C-G/C-G | 25 (.054) | 33 (.072) | .73 | .279 | 23 (.046) | 34 (.068) | .66 | .172 | 28 (.058) | 25 (.059) | .99 | 1 | .78 (.57–1.06) | .415 |
| X/X–C-G/X | 10 (.021) | 25 (.054) | .38 | .009 | 10 (.020) | 20 (.040) | .49 | .094 | 8 (.017) | 20 (.047) | .34 | .011 | .40 (.25–.61) | 7.42×10−4 |
| X/X–X/X | 3 (.006) | 1 (.002) | 2.97 | .624 | 1 (.002) | 3 (.006) | .33 | .624 | 0 | 2 (.005) | .00 | .219 | … | … |
Because of small counts in some of the cells, as well as the distribution of these counts, global P values were obtained by performing a permutation procedure on the data and generating a log-likelihood ratio homogeneity statistic for each permutation. The convergence of the statistic to its limiting distribution was measured by plotting the first two central moments as a function of the number of replicates. When the number of replicates reached a number in which the error of the P value estimates from a subsequent modeling procedure was negligible (<1%), a gamma distribution was fit using maximum-likelihood estimates for the parameters. Integration of the resulting gamma distribution yielded the global P values.
Calculated using Fisher’s combined test.
Allele 1 rs3212227-allele 1 rs6887695/allele 2 rs3212227-allele 2 rs6887695–allele 1 rs7530511-allele 1 rs11209026/allele 2 rs7530511-allele 2 rs11209026. For this analysis, the three nonrisk haplotypes for each gene were combined and were termed “X.”
Calculated using Fisher’s exact test.
Calculated using a Mantel-Haenszel common OR.
Calculated for diplotypes with the same effect (risk or protection) in all three sample sets, with use of Fisher’s combined test.
Discussion
We report the identification of two psoriasis-susceptibility genes, IL12B and IL23R, in whites of European descent. These genes were identified using a collection of >25,000 primarily functional SNPs evaluated in three independent case-control sample sets (1,446 patients and 1,432 controls) in a multistaged strategy that combined pooled and individual genotyping, along with single-marker and multimarker analyses. This approach, which was designed to increase power while simultaneously making effective use of genotyping resources and DNA, has been successfully used to identify genetic variants associated with multiple common, complex diseases, including rheumatoid arthritis,48 myocardial infarction,16,49 liver fibrosis,50 Alzheimer disease,51 and now psoriasis. Some of the genes identified in these studies do not colocalize with known disease-specific linkage peaks. Interestingly, IL23R (1p31.3) maps within the broad PSORS7 1p linkage peak defined by Veal and colleagues,52 whereas IL12B (5q31.1-q33.1) does not map to any of the seven other major PSORS linkage peaks reviewed by Bowcock and Kreuger.5 These data suggest that our multitiered, genecentric case-control approach is a highly effective strategy for identifying markers associated with common, complex diseases that complements traditional linkage studies.
Our IL12B findings confirm and extend the observation of Tsunemi et al.,13 who reported association of rs3212227 with risk of psoriasis in a Japanese study of 143 patients and 100 controls (f(A)=0.601 in cases vs. 0.505 in controls, OR 1.47, P=.035). Although the frequency of the risk allele is quite different in Japanese (∼50.5%) compared with white North American (∼78.5%) controls, this allele exhibits a similar modest risk of psoriasis in both populations (OR 1.47 in Japanese vs. ORcommon 1.56 in white North Americans). The decreased allele frequency of the IL12B susceptibility allele in Japanese individuals relative to white North American individuals parallels the difference in psoriasis prevalence in these two populations (1% in Japanese vs. 2%–3% in white North Americans).53 No other IL12B-associated SNPs were interrogated in the Japanese study.
Although IL12B encodes the common IL12-p40 subunit of both IL-12 and IL-23, IL-23 was discovered a decade after IL-12. Consequently, early studies of IL-12 function, which used either anti–IL-12p40 antibodies or IL-12p40–deficient mice, attributed functions to IL-12 that are only now being correctly ascribed to IL-23. Although this work is still ongoing, it is clear that each cytokine has a unique role in the immune response. Both are produced by activated dendritic cells and macrophages; however, regulation of their expression differs both spatially and temporally. In addition, they drive the development of distinct subsets of T-cells. IL-12 induces the classic interferon-γ–producing T-helper (Th) 1 cells, whereas IL-23 drives the expansion and maintenance of the newly defined ThIL-17 T cells (for a review, see the work of Hunter54 and Bowman et al.55).
Given that IL-12 drives the development of Th1 cells, which play an important role in a subset of T-cell–mediated autoimmune diseases, IL-12 and, more importantly, rs3212227 have been studied in a number of immune-mediated diseases. In addition to psoriasis, the A allele at this SNP has been associated with type 1 diabetes,56 and the C allele has been associated with diseases that can be classified as type 2 diseases (those dominated by a humoral response), such as asthma,57 atopic dermatitis,13 and chronic hepatitis C virus infection.58 However, with the exception of psoriasis, most of these reported disease associations have not been replicated in independent studies. Other diseases, such as rheumatoid arthritis59,60 (A. B. Begovich, S. J. Schrodi, and M. Chang, unpublished data), Crohn disease,61,62 and systemic lupus erythematosus,63 have not shown association with either allele at rs3212227.
Although the effect of the 3′ UTR rs3212227 SNP on IL-12 and IL-12p40 expression levels has been studied by multiple groups, the results are inconsistent. When compared with the C allele of this 3′ UTR SNP, the A allele has been reported to be associated with (i) increased IL-12p40 secretion,64 (ii) no change in IL-12p40 secretion but decreased IL-12 secretion,61 (iii) increased IL12B mRNA,56 and (iv) decreased IL-12 secretion.47 However, different cell types and methods of cell stimulation and IL-12 quantitation were used for these studies. To resolve this issue, additional studies are required, and they should involve specific cell types that are well characterized for the relevant genetic markers.
The conflicting results reported for the functional consequences of the rs3212227 SNP could be explained if several polymorphisms were working together synergistically or antagonistically to affect IL12B mRNA and protein levels or if an unidentified polymorphism at the IL12B locus that is in LD with rs3212227 was responsible for the psoriasis-associated risk at this locus. Both of these explanations are consistent with our observation that a second SNP upstream of IL12B, rs6887695, independently contributes to psoriasis risk and that together these two SNPs mark a set of haplotypes with a complex hierarchy of psoriasis risk. To address this issue further, we are currently conducting a detailed analysis of SNPs in high-to-moderate LD with both disease-associated IL12B SNPs, to complement the tag SNP and functional SNP approach described in this article.
Both IL-12 and IL-23 have been proposed as targets for the treatment of psoriasis. IL-12p40 antagonists have been used for the effective treatment of a number of animal models of inflammatory-based diseases, including a mouse model of psoriasis,65 and monoclonal antibodies directed against the IL-12p40 subunit are now in clinical trials for Crohn disease, multiple sclerosis, and psoriasis.66–68 Preliminary results show one of these biologics to be highly effective for psoriasis treatment.67 However, given that IL12B encodes the common subunit of two functionally distinct cytokines, IL-12 and IL-23,14,54 this antibody effectively functions as a broad-spectrum immune modulator, leading some to suggest that targeting only one of these cytokine pathways may be a safer, yet equally effective, therapy.55
Biological data suggest that the IL-23 pathway may be an important target for intervention in psoriasis. Patients with psoriasis have significantly increased levels of IL23A and IL12B mRNA but not IL12A mRNA in psoriatic lesions compared with nonlesional skin, as determined by quantitative RT-PCR69 and confirmed by immunohistochemical staining.70 Moreover, in a transgenic mouse model that overexpressed IL-12p40, IL-23 but not IL-12 was observed to be constitutively expressed by basal keratinocytes, which are thought to play a pivotal role in psoriasis pathobiology.71 The results of a recent study reporting that intradermal administration of IL-23 but not of IL-12 in mouse skin initiates a tumor necrosis factor–dependent epidermal hyperplasia and psoriasform lesions supports the notion that IL-23 dysregulation is likely to play an important role in psoriasis pathogenesis.72
Our finding that common variants in both IL12B and IL23R are associated with psoriasis risk provides genetic evidence that the IL-23 pathway may be an appropriate target for intervention in psoriasis. Given that IL23R lies just upstream of the gene encoding the IL12-receptor–specific protein, IL12RB2, detailed fine mapping of this region is currently under way to determine which gene or genes directly contribute to psoriasis risk. However, review of the CEU HapMap phase II data42 provides no evidence of LD between rs7530511 and any IL12RB2 SNPs and evidence of only modest LD between rs1120926 and a single IL12RB2 intronic SNP, rs2001257 (r2=0.253).
Duerr and colleagues73 recently reported strong association between one of the psoriasis-associated IL23R SNPs (rs11209026, Q381R) described here and risk of inflammatory bowel disease. No association was observed with the other IL23R SNP described here (rs7530511, L310P), and haplotypes were not evaluated. Together, these data are intriguing and add further support to the hypothesis that there are common genetic variants that contribute to general immune dysregulation and susceptibility to autoimmunity.74,75 Given the role of IL-23 and IL-12 in murine experimental autoimmune encephalomyelitis,76,77 these IL12B and IL23R variants merit evaluation in studies of multiple sclerosis as well as other autoimmune diseases.
In conclusion, we provide convincing statistical support for two psoriasis-susceptibility loci—one in 5q31.1-q33.1 (IL12B region) and the other in 1p31.3 (IL23R region)—and preliminary analyses suggest that the combination of risk and protective haplotypes at both loci can lead to a more-than-threefold differential risk for disease. These data provide additional justification for targeting both the IL-12 and IL-23 pathways with new psoriasis therapeutics and suggest that targeting IL-23 or downstream effector cytokines may directly target the disease pathway and prove efficacious for the treatment of psoriasis. Finally, it will be important to determine whether any of the identified IL12B and IL23R psoriasis-associated alleles are also associated with response to anti–IL-12p40 therapy and/or the most effective dosage of this antibody.
Acknowledgments
We are grateful to all the individuals with psoriasis and to the control individuals, for their participation in this study; to members of the Celera High Throughput and Computational Biology groups, for their support; to D. Lew and T. Vess, for the genotyping validation data; to M. McGinnis and J. Capper of Atria Genetics, for HLA-C genotyping; to C. Rowland, for very helpful statistical genetics comments; to A. Grupe, for discussions on DNA pooling strategies; to J. Lemaire and S. Mahan, for database and sample management at GCI; to A. Peiffer and M. Dixon, for invaluable input; to M. Hoffman, T. Christensen, T. Nelson, and B. Wong, for help recruiting patients and coordinating the project at the University of Utah; to M. Paul and everyone at LineaGen, for managing the collaboration; and to J. Sninsky, S. Broder, L. Honigberg, and E. Beasley, for insightful comments on this study. This study was supported in part by a Public Health Services research grant to the Huntsman General Clinical Research Center at the University of Utah, by National Center for Research Resources grant MO1-RR00064, and by generous gifts from the W. M. Keck Foundation and from the George S. and Delores Doré Eccles Foundation.
Web Resources
Accession numbers and URLs for data presented herein are as follows:
- dbSNP, http://www.ncbi.nlm.nih.gov/SNP/ (for SNPs rs10035989, rs1109918, rs11171806, rs11209026, rs11575926, rs11744690, rs1368437, rs1422668, rs1422878, rs1433048, rs17860508,rs1833754, rs1884444, rs1897565, rs2043270, rs2161357,rs2227314, rs2243131, rs2243149, rs2243154, rs2371494,rs270654, rs270661, rs2914119, rs3212220, rs3212227, rs3213096, rs3213119, rs375947, rs393548, rs401502, rs4921226, rs4921496, rs583911, rs6556398, rs6771983, rs6869411, rs6887695, rs6896438, rs6897374, rs6898290, rs717925, rs7530511, rs7709212, rs7721001, rs918520, rs929779, rs953861, and ss52085990)
- Entrez Nucleotide, http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?CMD=search&DB=nucleotide (for 5,000 bp upstream of the 5′-most exon of IL12B [accession number NW_922784] and chromosome 5 genomic contig [accession number NT_023133])
- Genetic Association Database, http://geneticassociationdb.nih.gov/
- HapMap, http://www.hapmap.org/
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for PSORS1, IL12B, IL23R, and mycobacterial disease)
- Primer3, http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi
- SeattleSNPs, http://pga.gs.washington.edu/
- SNPAnalyzer, http://snp.istech.info/
References
- 1.Lebwohl M (2003) Psoriasis. Lancet 361:1197–1204 10.1016/S0140-6736(03)12954-6 [DOI] [PubMed] [Google Scholar]
- 2.Bowcock AM (2005) The genetics of psoriasis and autoimmunity. Annu Rev Genomics Hum Genet 6:93–122 10.1146/annurev.genom.6.080604.162324 [DOI] [PubMed] [Google Scholar]
- 3.Gladman DD, Antoni C, Mease P, Clegg DO, Nash P (2005) Psoriasis arthritis: epidemiology, clinical features, course, and outcome. Ann Rheum Dis 64:ii14–ii17 10.1136/ard.2004.032482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhalerao J, Bowcock AM (1998) The genetics of psoriasis: a complex disorder of the skin and immune system. Hum Mol Genet 7:1537–1545 10.1093/hmg/7.10.1537 [DOI] [PubMed] [Google Scholar]
- 5.Bowcock AM, Krueger JG (2005) Getting under the skin: the immunogenetics of psoriasis. Nat Rev Immunol 5:699–711 10.1038/nri1689 [DOI] [PubMed] [Google Scholar]
- 6.Helms C, Saccone NL, Cao L, Daw JA, Cao K, Hsu TM, Taillon-Miller P, Duan S, Gordon D, Pierce B, et al (2005) Localization of PSORS1 to a haplotype block harboring HLA-C and distinct from corneodesmosin and HCR. Hum Genet 118:466–476 10.1007/s00439-005-0048-2 [DOI] [PubMed] [Google Scholar]
- 7.Nair RP, Stuart PE, Nistor I, Hiremagalore R, Chia NV, Jenisch S, Weichenthal M, Abecasis GR, Lim HW, Christophers E, et al (2006) Sequence and haplotype analysis supports HLA-C as the psoriasis susceptibility 1 gene. Am J Hum Genet 78:827–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Helms C, Cao L, Krueger JG, Wijsman EM, Chamian F, Gordon D, Heffernan M, Daw JA, Robarge J, Ott J, et al (2003) A putative RUNX1 binding site variant between SLC9A3R1 and NAT9 is associated with susceptibility to psoriasis. Nat Genet 35:349–356 10.1038/ng1268 [DOI] [PubMed] [Google Scholar]
- 9.Hewett D, Samuelsson L, Polding J, Enlund F, Smart D, Cantone K, See CG, Chadha S, Inerot A, Enerback C, et al (2002) Identification of a psoriasis susceptibility candidate gene by linkage disequilibrium mapping with a localized single nucleotide polymorphism map. Genomics 79:305–314 10.1006/geno.2002.6720 [DOI] [PubMed] [Google Scholar]
- 10.Hirschhorn JN, Daly MJ (2005) Genome-wide association studies for common diseases and complex traits. Nat Rev Genet 6:95–108 10.1038/nrg1521 [DOI] [PubMed] [Google Scholar]
- 11.Long AD, Langley CH (1999) The power of association studies to detect the contribution of candidate genetic loci to variation in complex traits. Genome Res 9:720–731 [PMC free article] [PubMed] [Google Scholar]
- 12.Risch N, Merikangas K (1996) The future of genetic studies of complex human diseases. Science 273:1516–1517 10.1126/science.273.5281.1516 [DOI] [PubMed] [Google Scholar]
- 13.Tsunemi Y, Saeki H, Nakamura K, Sekiya T, Hirai K, Fujita H, Asano N, Kishimoto M, Tanida Y, Kakinuma T, et al (2002) Interleukin-12 p40 gene (IL12B) 3′-untranslated region polymorphism is associated with susceptibility to atopic dermatitis and psoriasis vulgaris. J Dermatol Sci 30:161–166 10.1016/S0923-1811(02)00072-5 [DOI] [PubMed] [Google Scholar]
- 14.Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, Vega F, Yu N, Wang J, Singh K, et al (2000) Novel p19 protein engages IL12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 13:715–725 10.1016/S1074-7613(00)00070-4 [DOI] [PubMed] [Google Scholar]
- 15.Germer S, Holland MJ, Higuchi R (2000) High-throughput SNP allele-frequency determination in pooled DNA samples by kinetic PCR. Genome Res 10:258–266 10.1101/gr.10.2.258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shiffman D, Ellis SG, Rowland CM, Malloy MJ, Luke MM, Iakoubova OA, Pullinger CR, Cassano J, Aouizerat BE, Fenwick RG, et al (2005) Identification of four gene variants associated with myocardial infarction. Am J Hum Genet 77:596–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bustamante CD, Fledel-Alon A, Williamson S, Nielsen R, Hubisz MT, Glanowski S, Tanenbaum DM, White TJ, Sninsky JJ, Hernandez RD, et al (2005) Natural selection on protein-coding genes in the human genome. Nature 437:1153–1157 10.1038/nature04240 [DOI] [PubMed] [Google Scholar]
- 18.Li Y, Tacey K, Doil L, van Luchene R, Garcia V, Rowland C, Schrodi S, Leong D, Lau K, Cantanese J, et al (2004) Association of ABCA1 with late-onset Alzheimer’s disease is not observed in a case-control study. Neurosci Lett 366:268–271 10.1016/j.neulet.2004.05.047 [DOI] [PubMed] [Google Scholar]
- 19.Carlton VEH, Hu X, Chokkalingam AP, Schrodi SJ, Brandon R, Alexander HC, Chang M, Catanese JJ, Leong DU, Ardlie KG, et al (2005) PTPN22 genetic variation: evidence for multiple variants associated with rheumatoid arthritis. Am J Hum Genet 77:567–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rozen S, Skaletsky H (2000) Primer3 on the WWW for general users and for biologist programmers. In: Krawetz S, Misener S (eds) Bioinformatics methods and protocols: methods in molecular biology. Humana Press, Totowa, NJ, pp 365–386 [DOI] [PubMed] [Google Scholar]
- 21.Hu X, Schrodi SJ, Ross DA, Cargill, M (2004) Selecting tagging SNPs for association studies using power calculations from genotype data. Hum Hered 57:156–170 10.1159/000079246 [DOI] [PubMed] [Google Scholar]
- 22.Abecasis GR, Cookson WO (2000) GOLD—graphical overview of linkage disequilibrium. Bioinformatics 16:182–183 10.1093/bioinformatics/16.2.182 [DOI] [PubMed] [Google Scholar]
- 23.Yoo J, Seo B, Kim Y (2005) SNPAnalyzer: a web-based integrated workbench for single-nucleotide polymorphism analysis. Nucleic Acids Res 33:W483–W488 10.1093/nar/gki428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schaid DJ, Rowland CM, Tines DE, Jacobson RM, Poland GA (2002) Score tests for association between traits and haplotypes when linkage phase is ambiguous. Am J Hum Genet 70:425–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fisher RA (1954) Statistical methods for research workers, 12th ed. Oliver and Boyd, Edinburgh [Google Scholar]
- 26.Sokal RR, Rohlf FJ (1995) Biometry, 3rd ed. W. H. Freeman and Company, New York [Google Scholar]
- 27.Weir BS (1996) Genetic data analysis II. Sinauer, Sunderland, MA [Google Scholar]
- 28.Nielsen DM, Ehm MG, Zaykin DV, Weir BS (2004) Effect of two- and three-locus linkage disequilibrium on the power to detect marker/phenotype associations. Genetics 168:1029–1040 10.1534/genetics.103.022335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Breslow NE, Day NE (1980) Statistical methods in cancer research. Vol I. The analysis of case-control studies. International Agency for Research on Cancer, Lyon [PubMed] [Google Scholar]
- 30.Walter SD (1975) The distribution of Levin’s measure of attributable risk. Biometrika 62:371–374 10.2307/2335374 [DOI] [Google Scholar]
- 31.Schlesselman JJ (1982) Case-control studies: design, conduct, analysis. Oxford University Press, New York [Google Scholar]
- 32.Schrodi SJ (2005) A probabilistic approach to large-scale association scans: a semi-Bayesian method to detect disease-predisposing alleles. Stat Appl Genet Mol Biol 4:Article31 [DOI] [PubMed] [Google Scholar]
- 33.Efron B, Tibshirani R, Storey JD, Tusher V (2001) Empirical Bayes analysis of a microarray experiment. J Am Stat Assoc 96:1151–1160 10.1198/016214501753382129 [DOI] [Google Scholar]
- 34.Devlin B, Roeder K (1999) Genomic control for association studies. Biometrics 55:997–1004 10.1111/j.0006-341X.1999.00997.x [DOI] [PubMed] [Google Scholar]
- 35.Thomson G, Robinson WP, Kuhner MK, Joe S, MacDonald MJ, Gottschall JL, Barbosa J, Rich SS, Bertrams J, Baur MP, et al (1988) Genetic heterogeneity, modes of inheritance, and risk estimates for a joint study of Caucasians with insulin-dependent diabetes mellitus. Am J Hum Genet 43:799–816 [PMC free article] [PubMed] [Google Scholar]
- 36.Valdes AM, Thomson G (1997) Detecting disease-predisposing variants: the haplotype method. Am J Hum Genet 60:703–716 [PMC free article] [PubMed] [Google Scholar]
- 37.Jeffreys H (1935) Some tests of significance, treated by the theory of probability. Proc Camb Philol Soc 31:203–222 [Google Scholar]
- 38.Kass R, Raftery A (1995) Bayes factors and model uncertainty. J Am Stat Assoc 90:773–795 10.2307/2291091 [DOI] [Google Scholar]
- 39.Ahmad T, Neville M, Marshall SE, Armuzzi A, Mulcahy-Hawes K, Crawshaw J, Sato H, Ling KL, Barnardo M, Goldthorpe S, et al (2003) Haplotype-specific linkage disequilibrium patterns define the genetic topography of the human MHC. Hum Mol Genet 12:647–656 10.1093/hmg/12.6.647 [DOI] [PubMed] [Google Scholar]
- 40.Altare F, Lammas D, Revy P, Jouanguy E, Doffinger R, Lamhamedi S, Drysdale P, Scheel-Toellner D, Girdlestone J, Darbyshire P, et al (1998) Inherited interleukin 12 deficiency in a child with bacille Calmette-Guerin and Salmonella enteritidis disseminated infection. J Clin Invest 102:2035–2040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Picard C, Fieschi C, Altare F, Al-Jumaah S, Al-Hajjar S, Feinberg J, Dupuis S, Soudais C, Al-Mohsen IZ, Genin E, et al (2002) Inherited interleukin-12 deficiency: IL12B genotype and clinical phenotype of 13 patients from six kindreds. Am J Hum Genet 70:336–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.The International HapMap Consortium (2005) A haplotype map of the human genome. Nature 437:1299–1320 10.1038/nature04226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang D, Cancilla MR, Morahan G (2000) Complete primary structure, chromosomal localisation, and definition of polymorphisms of the gene encoding the human interleukin-12 p40 subunit. Genes Immun 1:515–520 10.1038/sj.gene.6363720 [DOI] [PubMed] [Google Scholar]
- 44.Morahan G, Huang D, Wu M, Holt BJ, White GP, Kendall GE, Sly PD, Holt PG (2002) Association of IL12B promoter polymorphism with severity of atopic and non-atopic asthma in children. Lancet 360:455–459 10.1016/S0140-6736(02)09676-9 [DOI] [PubMed] [Google Scholar]
- 45.Litjens NH, van der Plas MJ, Ravensbergen B, Numan-Ruberg SC, van Assen Y, Thio HB, van Dissel JT, van de Voss E, Nibbering PH (2004) Psoriasis is not associated with IL-12p70/IL-12p40 production and IL12B promoter polymorphism. J Invest Dermatol 122:923–926 10.1111/j.0022-202X.2004.22427.x [DOI] [PubMed] [Google Scholar]
- 46.Muller-Berghaus J, Kern K, Paschen A, Nguyen XD, Kluter H, Morahan G, Schadendorf D (2004) Deficient IL-12p70 secretion by dendritic cells based on IL12B promoter genotype. Genes Immun 5:431–434 10.1038/sj.gene.6364102 [DOI] [PubMed] [Google Scholar]
- 47.Yilmaz V, Yentur SP, Saruhan-Direskeneli G (2005) IL-12 and IL-10 polymorphisms and their effects on cytokine production. Cytokine 30:188–194 10.1016/j.cyto.2005.01.006 [DOI] [PubMed] [Google Scholar]
- 48.Begovich AB, Carlton VEH, Honigberg LA, Schrodi SJ, Chokkalingam AP, Alexander HC, Ardlie KG, Qiqing H, Smith AM, Spoerke JM, et al (2004) A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am J Hum Genet 75:330–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shiffman D, Rowland CM, Louie JZ, Luke MM, Bare LA, Bolonick JI, Young BA, Catanese JJ, Stiggins CF, Pullinger CR, et al (2006) Gene variants of VAMP8 and HNRPUL1 are associated with early-onset myocardial infarction. Arterioscler Thromb Vasc Biol 26:1613–1618 10.1161/01.ATV.0000226543.77214.e4 [DOI] [PubMed] [Google Scholar]
- 50.Huang H, Shiffman ML, Cheung RC, Layden TJ, Friedman S, Abar OT, Yee L, Chokkalingam AP, Schrodi SJ, Chan J, et al (2006) Identification of two gene variants associated with risk of advanced fibrosis in patients with chronic hepatitis C. Gastroenterology 130:1679–1687 10.1053/j.gastro.2006.02.032 [DOI] [PubMed] [Google Scholar]
- 51.Li Y, Grupe A, Rowland C, Nowotny P, Kauwe JS, Smemo S, Hinrichs A, Tacey K, Toombs TA, Kwok S, et al (2006) DAPK1 variants are associated with Alzheimer’s disease and allele-specific expression. Hum Mol Genet 15:2560–2568 10.1093/hmg/ddl178 [DOI] [PubMed] [Google Scholar]
- 52.Veal CD, Clough RL, Barber RC, Mason S, Tillman D, Ferry B, Jones AB, Ameen M, Balendran N, Powis SH, et al (2001) Identification of a novel psoriasis susceptibility locus at 1p and evidence of epistasis between PSORS1 and candidate loci. J Med Genet 38:7–13 10.1136/jmg.38.1.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yip SY (1984) The prevalence of psoriasis in the Mongoloid race. J Am Acad Dermatol 10:965–968 [DOI] [PubMed] [Google Scholar]
- 54.Hunter CA (2005) New IL-12-family members: IL-23 and IL-27, cytokines with divergent functions. Nat Rev Immunol 5:521–531 10.1038/nri1648 [DOI] [PubMed] [Google Scholar]
- 55.Bowman EP, Chackerian AA, Cua DJ (2006) Rationale and safety of anti-interleukin-23 and anti-interleukin-17A therapy. Curr Opin Infect Dis 19:245–252 10.1097/01.qco.0000224818.42729.67 [DOI] [PubMed] [Google Scholar]
- 56.Morahan G, Huang D, Ymer SI, Cancilla MR, Stephen K, Dabadghao P, Werther G, Tait BD, Harrison LC, Colman PG (2001) Linkage disequilibrium of a type 1 diabetes susceptibility locus with a regulatory IL12B allele. Nat Genet 27:218–221 10.1038/84872 [DOI] [PubMed] [Google Scholar]
- 57.Randolph AG, Lange C, Silverman EK, Lazarus R, Silverman ES, Raby B, Brown A, Ozonoff A, Richter B, Weiss ST (2004) The IL12B gene is associated with asthma. Am J Hum Genet 75:709–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mueller T, Mas-Marques A, Sarrazin C, Wiese M, Halangk J, Witt H, Ahlenstiel G, Spengler U, Goebel U, Wiedenmann B, et al (2004) Influence of interleukin 12B (IL12B) polymorphisms on spontaneous and treatment-induced recovery from hepatitis C virus infection. J Hepatol 41:652–658 10.1016/j.jhep.2004.06.021 [DOI] [PubMed] [Google Scholar]
- 59.Hall MA, McGlinn E, Coakley G, Fisher SA, Boki K, Middleton D, Kaklamani E, Moutsopoulos H, Loughran TP Jr, Ollier WE, et al (2000) Genetic polymorphism of IL-12 p40 gene in immune-mediated disease. Genes Immun 1:219–224 10.1038/sj.gene.6363661 [DOI] [PubMed] [Google Scholar]
- 60.Orozco G, Gonzalez-Gay MA, Paco L, Lopez-Nevot MA, Guzman M, Pascual-Salcedo D, Balsa A, Martin J (2005) Interleukin 12 (IL12B) and interleukin 12 receptor (IL12RB1) gene polymorphisms in rheumatoid arthritis. Hum Immunol 66:710–715 10.1016/j.humimm.2005.02.004 [DOI] [PubMed] [Google Scholar]
- 61.Seegers D, Zwiers A, Strober W, Pena AS, Bouma G (2002) A TaqI polymorphism in the 3′UTR of the IL-12 p40 gene correlates with increased IL-12 secretion. Genes Immun 3:419–423 10.1038/sj.gene.6363919 [DOI] [PubMed] [Google Scholar]
- 62.Zwiers A, Seegers D, Heijmans R, Koch A, Hampe J, Nikolaus S, Pena AS, Schreiber S, Bouma G (2004) Definition of polymorphisms and haplotypes in the interleukin-12B gene: association with IL-12 production but not with Crohn’s disease. Genes Immun 5:675–677 10.1038/sj.gene.6364131 [DOI] [PubMed] [Google Scholar]
- 63.Sanchez E, Morales S, Paco L, Lopez-Nevot MA, Hidalgo C, Jimenez-Alonso J, Torres B, Gonzalez-Gay MA, Callejas JL, Ortego-Centeno N, et al (2005) Interleukin 12 (IL12B), interleukin 12 receptor (IL12RB1) and interleukin 23 (IL23A) gene polymorphism in systemic lupus erythematosus. Rheumatology 44:1136–1139 10.1093/rheumatology/keh697 [DOI] [PubMed] [Google Scholar]
- 64.Stanilova S, Miteva L (2005) Taq-I polymorphism in 3′UTR of the IL-12B and association with IL-12p40 production from human PBMC. Genes Immun 6:364–366 10.1038/sj.gene.6364213 [DOI] [PubMed] [Google Scholar]
- 65.Hong K, Chu A, Ludviksson BR, Berg EL, Ehrhardt RO (1999) IL-12, independently of IFN-γ, plays a crucial role in the pathogenesis of a murine psoriasis-like skin disorder. J Immunol 162:7480–7491 [PubMed] [Google Scholar]
- 66.Winterfield LS, Menter A, Gordon K, Gottlieb A (2005) Psoriasis treatment: current and emerging directed therapies. Ann Rheum Dis 64:ii87–ii90 10.1136/ard.2004.032276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kauffman CL, Aria N, Toichi E, McCormick TS, Cooper KD, Gottlieb AB, Everitt DE, Frederick B, Zhu Y, Graham MA, et al (2004) A phase I study evaluating the safety, pharmacokinetics, and clinical response of a human IL-12 p40 antibody in subjects with plaque psoriasis. J Invest Dermatol 123:1037–1044 10.1111/j.0022-202X.2004.23448.x [DOI] [PubMed] [Google Scholar]
- 68.Mannon PJ, Fuss IJ, Mayer L, Elson CO, Sandborn WJ, Present D, Dolin B, Goodman N, Groden C, Hornung RL, et al (2004) Anti-interleukin-12 antibody for active Crohn’s disease. N Engl J Med 351:2069–2079 10.1056/NEJMoa033402 [DOI] [PubMed] [Google Scholar]
- 69.Lee E, Trepicchio WL, Oestreicher JL, Pittman D, Wang F, Chamian F, Dhodapkar M, Krueger JG (2004) Increased expression of interleukin 23 p19 and p40 in lesional skin of patients with psoriasis vulgaris. J Exp Med 199:125–130 10.1084/jem.20030451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Piskin G, Sylva-Steenland RM, Bos JD, Teunissen MB (2006) In vitro and in situ expression of IL-23 by keratinocytes in healthy skin and psoriasis lesions: enhanced expression in psoriatic skin. J Immunol 176:1908–1915 [DOI] [PubMed] [Google Scholar]
- 71.Kopp T, Lenz P, Bello-Fernandez C, Kastelein RA, Kupper TS, Stingl G (2003) IL-23 production by cosecretion of endogenous p19 and transgenic p40 in keratin 14/p40 transgenic mice: evidence for enhanced cutaneous immunity. J Immunol 170:5438–5444 [DOI] [PubMed] [Google Scholar]
- 72.Chan JR, Blumenschein W, Murphy E, Diveu C, Wiekowski M, Abbondanzo S, Lucian L, Geissler R, Brodie S, Kimball AB, et al (2006) IL-23 stimulates epidermal hyperplasia via TNF and IL-20R2-dependent mechanisms with implications for psoriasis pathogenesis. J Exp Med 203:2577–2587 10.1084/jem.20060244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, Steinhart AH, Abraham C, Regueiro M, Griffiths A, et al (2006) A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science 314:1461–1463 10.1126/science.1135245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Marrack P, Kappler J, Kotzin BL (2001) Autoimmune disease: why and where it occurs. Nat Med 7:899–905 10.1038/90935 [DOI] [PubMed] [Google Scholar]
- 75.Wandstrat A, Wakeland E (2001) The genetics of complex autoimmune disease: non-MHC susceptibility genes. Nat Immunol 2:802–809 10.1038/ni0901-802 [DOI] [PubMed] [Google Scholar]
- 76.Becher B, Durell BG, Noelle RJ (2002) Experimental autoimmune encephalitis and inflammation in the absence of interleukin-12. J Clin Invest 110:493–498 10.1172/JCI200215751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, et al (2003) Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 421:744–748 10.1038/nature01355 [DOI] [PubMed] [Google Scholar]