
Abstract
Abalone (family Haliotidae) are an ecologically and economically significant group of marine gastropods that can be found in tropical and temperate waters. To date, only a few Haliotis genomes are available, all belonging to temperate species. Here, we provide the first chromosome-scale abalone genome assembly and the first reference genome of the tropical abalone Haliotis asinina. The combination of PacBio long-read HiFi sequencing and Dovetail’s Omni-C sequencing allowed the chromosome-level assembly of this genome, while PacBio Isoform sequencing across five tissue types enabled the construction of high-quality gene models. This assembly resulted in 16 pseudo-chromosomes spanning over 1.12 Gb (98.1% of total scaffolds length), N50 of 67.09 Mb, the longest scaffold length of 105.96 Mb, and a BUSCO completeness score of 97.6%. This study identified 25,422 protein-coding genes and 61,149 transcripts. In an era of climate change and ocean warming, this genome of a heat-tolerant species can be used for comparative genomics with a focus on thermal resistance. This high-quality reference genome of H. asinina is a valuable resource for aquaculture, fisheries, and ecological studies.
Similar content being viewed by others


Background & Summary
Abalone (Haliotis) are a genus of marine herbivorous gastropods found in tropical and temperate coastal waters on every continent except for the Pacific coast of South America and the Atlantic coast of North America1. In addition to their ecological, historical, and cultural importance2,3,4, abalone are a highly prized seafood product that underpins valuable wild-harvest and aquaculture industries in many countries5,6. There has been a significant decrease in wild populations of abalone largely due to illegal harvesting, pollution, climate change and disease5,7,8. As a result, many species of abalone are recognized to be at risk – the IUCN Red ListTM lists 44% of abalone species as being threatened with extinction9,10.
Whether in the wild or in aquaculture, abalone are also at risk due to ocean warming and extreme environmental events11,12. In the summer of 2011, between early February and early March, wild Roe’s abalone stocks suffered significant, if not total, mortality around Kalbarri, Western Australia13. Similarly, the 2016 mortality event of wild abalone near the coast of Tasmania, Australia, led to smaller catches and reduced quotas14. These events have great economic impacts on abalone fisheries, resulting in a significant decrease in production and loss of income. The increase in abalone aquaculture and the concerns for wild populations worldwide have motivated researchers to apply omics tools to provide genetic resources, improve knowledge regarding this genus, and ultimately aid production and conservation. To date, the great majority of the genetic resources available for abalone are of temperate species15,16,17,18,19,20,21. No reference genome for any tropical abalone species has been published to date.
The Donkey’s ear abalone, Haliotis asinina (Linnaeus, 1758), is the largest of the tropical abalone species. It is also the fastest-growing abalone of all abalone species22. This species is distributed throughout the Indo-Pacific and is highly desired as seafood, mainly in South-East Asia23,24. Due to its popularity, wild stocks are at risk as a result of overfishing25. Efforts are underway to revive H. asinina populations through stock enhancement and the use of marine reserves26. The lack of genetic data available for this species limits studies on genetic variation (between and within abalone species), development of genetic breeding programs, connectivity and genetic technologies that will assist fisheries, aquaculture and conservation strategies.
Here, we provide the first reference genome of the tropical abalone H. asinina. Furthermore, this is the first chromosome-scale genome assembly of any abalone species (to date). Using Pacific Biosciences of California, Inc. (PacBio) 5-base HiFi sequencing, Dovetail Genomics Omni-C approach and PacBio Isoform sequencing (Iso-Seq), we assembled and annotated the 1.14 Gb length reference genome. The total genome length was assembled into 170 scaffolds, with an N50 of 67.09 Mb, L90 of 15, a BUSCO completeness score of 97.6% and a k-mer completeness of 99.5%. Over 98% of the scaffold’s length was anchored to 16 pseudo-chromosomes. The chromosome number matches the findings of the previous karyotype studies27. Furthermore, 40.0% of the genome was identified as repetitive sequences. A total of 25,422 protein-coding genes were predicted, including 61,149 transcripts. In addition, we used the same data to measure DNA methylation across the genome and to assemble the mitochondrial genome of H. asinina.
This significant resource, along with the use of omics tools (i.e., comparative genomics, transcriptomic, epigenomics and proteomics), will provide new insights regarding the evolution of abalone and genetic factors that might assist in overcoming the current and future challenges mentioned above.
Methods
The general workflow is illustrated in Fig. 1.

Schematic overview of the study workflow.
Biological materials
In April 2022, H. asinina individuals were obtained from Arlington Reef (−16° 42′ 26.1036″S, 146° 3′ 30.4128″E) on the Great Barrier Reef, Australia, by divers from Cairns Marine Pty Ltd. Abalone were introduced into a round 100 L white plastic aquaria at the Marine and Aquaculture Research Facility (MARF) at James Cook University (Townsville, Australia). High water quality was maintained during the entire period. The temperature was set to the ambient temperatures at the collection site and was recorded continuously using the facility’s automated monitoring system. Water quality parameters, including ammonia, nitrate, and nitrite, were measured using “AquaSonic” kits. Water in the aquaria was replaced every two to three days. The abalone were fed every two days using Halo abalone feed (3 mm pellets) manufactured by Skretting.
Sampling, nucleic acid extraction, library preparation and sequencing
Sampling, nucleic acid extraction, library preparation and sequencing were all performed on the same individual (described below).
Following a fasting period of 24-hours, one abalone individual (female, body length = 10.9 cm, shell length = 7.4 cm) was randomly selected and dissected immediately for High Molecular Weight Genomic DNA (HMW gDNA) extraction. HMW gDNA was extracted from the ~30 mg of fresh muscle tissue using the Circulomics® Nanobind Tissue Big DNA Kit following protocol modification for Aplysia28. Library preparation and sequencing were performed by the Australian Genome Research Facility (AGRF) according to PacBio protocols. Sequencing was performed using a single SMRT Cell and the PacBio Sequel ΙΙ (specifically, 5-base HiFi sequencing) with seq polymerase version 2.2 and seq primer v5. Movie time was 30hrs and 120pM SMRTcell loading. This resulted in 36.2 GB of data with 2.62 M (million) high-quality reads (Table 1).
The DNase Hi-C (Omni-C) library was prepared using the Dovetail Omni-C® Kit at AGRF according to the manufacturer’s protocol with modifications as follows: 60 mg of abalone muscle tissue was thoroughly cryo-ground using liquid nitrogen, and the chromatin was fixed with disuccinimidyl glutarate (DSG) and formaldehyde in the nucleus. After removing the cross-linking reagents, the disrupted tissue sample underwent sequential filtration through 200 μm and 50 μm cell strainers to eliminate large debris. The cross-linked chromatin was then digested in situ with the optimal amount of DNase I to achieve efficient chromatin digestion and, hence, generate long-range cis reads. Following digestion, the cells were lysed with sodium dodecyl-sulfate (SDS) to extract the chromatin fragments. Stage 3 of the library preparation - proximity ligation, was optimised (1) by reducing the recommended input lysate, thereby minimising any impurities, and (2) by increasing the intra-aggregate bridge ligation to an overnight reaction to enhance the ligation events. Briefly, optimally digested chromatin fragments were bound to Chromatin Capture Beads. Next, the chromatin ends were repaired and ligated to a biotinylated bridge adapter, followed by proximity ligation of adapter-containing ends. After proximity ligation, the crosslinks were reversed, the associated proteins were degraded, and the DNA was purified and then converted into a sequencing library using Illumina-compatible adaptors. Biotin-containing fragments were isolated using streptavidin beads prior to PCR amplification. The library was sequenced on an Illumina Novaseq X plus a platform to generate two million 2 × 150 base-pairs (bp) read pairs to assess the quality of mapping, valid cis-trans reads and complexity of the library. For chromosome-level assembly, the Omni -C library was sequenced to achieve approximately 100 M 2 × 150 bp read pairs per GB of the genome size. This resulted in 144.43 GB of data, including 478.26 M reads (Table 1).
Total RNA was extracted from five tissue types: gonad, liver, epipodial tentacle, eyes and gills. Unfortunately, attempts to extract high-quality RNA from the muscle tissue were unsuccessful. Each tissue was crushed with a sterilized, chilled pestle and mortar using 1 ml of TRIzol™. Once the tissue disruption was completed, the lysate was kept at −20 °C overnight. Total RNA extraction was completed using TRIzol™ Plus RNA Purification Kit (Invitrogen™) following the manufacturer’s protocol. The extracted RNA was stored at −80 °C. Library prep and sequencing were performed by AGRF following PacBio protocols. Sequencing was done using the PacBio Sequel ΙΙ and yielded 5.90 GB of data with 3.13 M reads (Table 1).
Genome assembly and scaffolding
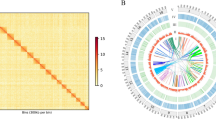
For the assembly, we used the Hifiasm version 0.19.729 haplotype-resolved de novo assembler with the PacBio HiFi adapter-free FASTA file and Hi-C partition using Omni-C data. Next, Omni-C data was used as input for Dovetail’s Omni-C workflow (https://github.com/dovetail-genomics/Omni-C). The workflow includes various tools30,31,32,33,34,35 for QC of the Omni-C library and generating contact maps. The primary assembly was indexed using SAMtools33 and the index file was used to generate the ‘.genome’ file. Omni-C reads were aligned to a reference genome using BWA version 0.7.1731, and high-quality mapped reads were retained. The mapped data was used as input for pairtools version 1.0.332 to identify proximity ligation events, categorize pairs by read type, insert distance, and flag and remove PCR duplicates. Juicer tools version 1.635 was used to generate the HiC contact matrix and contact map. The final scaffolding step was done using YaHs version 1.136, which resulted in 170 scaffolds that span over 1.14 Gb with the longest scaffold size of 105.96 Mb, N50 of 67.09 Mb and L90 of 15 (Table 2). The Hi-C map (Fig. 2) suggested 16 chromosome-scale scaffolds, comprising 98.1% of the total genome size (Table 3).

Hi-C heatmap. Pairwise interactions between pairs of chromosomes throughout H. asinina genome assembly. The figure was generated using Juicer35.
Methylation calling
High-quality reads produced using PacBio 5-base HiFi sequencing were used for CpG methylation calling across the genome assembly. Primrose version 1.3.0 (https://github.com/mattoslmp/primrose), a tool that predicts 5-methylcytosine (5mC) in HiFI reads, was used to add MM and ML tags (SAM tags that represent base modifications/methylation and base modification probabilities, respectively). The reads, which included the MM and ML tags, were aligned to the assembly using pbmm2 version 1.13.1 (https://github.com/PacificBiosciences/pbmm2), a minimap237,38 SMRT wrapper for PacBio data. Then, the aligned_bam_to_cpg_scores tool provided in pb-CpG-tools version 2.3.2 (https://github.com/PacificBiosciences/pb-CpG-tools) was used to generate CpG site methylation probabilities. Then, high probability (>95%) methylation site density was calculated across the entire genome (Fig. 3).

Chromosome ideogram. Each pair of ideograms represents one of the sixteen chromosomes in the H. asinina genome. The numbers at the bottom of each ideogram represent the number of the chromosome (i.e. 1 = chr1). The inner heatmap in the left ideogram of each pair represents the gene density (bin size = 0.5 Mb). The inner heatmap in the right ideogram of each pair represents the methylation density (bin size = 0.5 Mb, probability cut-off of 95%). The top colour scale represents the gene density, and the lower scale represents the methylation density. The figure was generated using RIdeogram79.
Mitochondrial genome assembly
MitoHiFi version 3.0.139, a pipeline for mitochondrial genome assembly from PacBio HiFi reads (or the assembled contigs/scaffolds), was used with the default annotation tools – MitoFinder version 1.4.140 and ARWEN41. Scaffold_159 corresponds to the mitochondrial genome (17450 bp in length), including all 37 identified mitochondrial genes, with no frameshifts and high probability (>96%).
Repetitive sequence identification
RepeatModeler version 2.0.542 and RepeatMasker version 4.1.543 were used to screen the H. asinina genome assembly for de novo identification of transposable elements (TEs) and classification of repeated and low complexity sequences (Table 4). The proportion of repeated elements in H. asinina genome was 38.42%, half of which were classified as unknown (19.71%). Retroelements (Class I) comprised 13.25%, DNA transposons (Class II) were 5.37%, and 1.30% were simple repeats. The proportion of repeats found in H. asinina genome is relatively similar to other abalone species16,17,19,21 and other marine invertebrates44,45 such as Aplysia californica46 and Crassostrea virginica47.
Gene prediction and functional annotation
Gene prediction was performed on a version of the genome that was soft-masked for repeats using RepeatMasker version 4.1.543. Then, the PacBio Secondary Analysis Tools on Bioconda37,48 were used to process the Iso-Seq reads and identify transcripts. Iso-Seq 3, a scalable de novo isoform discovery from single-molecule PacBio reads workflow was applied on the reads from all five tissue types (liver, gonad, eyes, gills and epipodial tentacle). The full workflow is detailed at https://github.com/ylipacbio/IsoSeq3. Briefly, cDNA primers, polyA tail and artificial concatemers were removed, and de novo isoform-level clustering was performed. High-quality isoforms were mapped to the genome (Fig. 4) using pbmm2 with a 99.86% mapping rate (samtools-flagstat version 1.16.133). Redundant transcripts were collapsed, and the TAMA49 package was used to produce gene models and to identify open reading frames (ORF) and coding regions (CDS). AGAT version 1.2.050 was used to filter all isoforms and to obtain the longest isoform per gene. For functional annotation, the protein-coding genes’ amino acid sequences were blasted (cut-off value 1e−5) using (1) blastp51,52 against UniProtKB/Swiss-Prot database53, (2) KEGG54,55, (3) InterProScan version Version 5.59-91.056,57 and (4) eggNOG version 2.1.858 to find protein hits, gene ontology and pathway information. Overall, 25,422 protein-coding genes and 61,149 transcripts were identified. The distribution and content of the gene elements are presented in Fig. 4. Gene density and methylation density across the 16 pseudo-chromosomes are presented in Fig. 3.

Circos plot of H. asinina genome characteristics. From the outer to the inner layer: (a) 16 chromosome-level scaffolds (values represent length in Mb), (b) GC content, (c) exon content, (d) 5′ untranslated region (UTR) content, (e) 3′ UTR content, (f) Iso-Seq mapping coverage and a photo of H. asinina female specimen that was used for this study. Generated and calculated using TBtools80 on the basis of 1 Mb windows.
Data Records
All sequencing data used in this study and the Whole Genome Shotgun (WGS) assembly have been submitted to the National Center for Biotechnology Information (NCBI) via BioProject ID PRJNA108003959. PacBio DNA sequencing data is available under the NCBI Sequence Read Archive accession number SRR2808376460. PacBio Iso-Seq data for all tissues (eyes, gills, tentacles, liver and gonad) is available under the NCBI Sequence Read Archive accession numbers SRR28084366-SRR2808437061,62,63,64,65. The Omni-C data is available under the NCBI Sequence Read Archive accession number SRR2810064366. The WGS assembly has been deposited at GenBank under the accession GCA_037392515.167. Genome annotation files68, repeat sequences files69 and the mitochondrial genome assembly70, genome methylation regions71 are available in Figshare.
Technical Validation
Nucleic acid
DNA quality and quantity was measured using Thermo Scientific™ NanoDrop (260/280 = 1.87; 260/230 = 2.13, 111.8 ng/ml) and Qubit dsDNA High Sensitivity Assay (106 ng/ml). The integrity of the HMW gDNA was also confirmed by the Australian Genome Research Facility (AGRF) using the Agilent™ FemtoPulse system. RNA quality and quantity from all tissues were measured using Thermo Scientific™ NanoDrop (260/280 = 2.07–2.14; 260/230 = 1.93–2.28) and the Agilent™ TapeStation 4150 system (RIN > 9.3).
Sequencing data, assembly and annotations
Using HiFiAdapterFilt version 2.0.072, the PacBio HiFi reads BAM file was converted into a FASTA file prior to the adapter filtering and read trimming (using the default settings). The adapter-free FASTA file was used for k-mer counting using Meryl version 1.473 with k = 20 (estimated with Meryl based on the genome size). Next, the k-mer database was used as input to estimate the overall characteristics of the genome (genome heterozygosity, repeat content, and size) from sequencing reads using a kmer-based statistical approach via GenomeScope 2.0 version 1.0.074,75 (Fig. 5). The Hifiasm primary assembly output was used as input for QUAST version 5.2.076 and Merqury version 1.373 to generate a quality assessment report of the assembly. We used BUSCO version 5.5.077 with the metazoan_odb10 database to assess the genome assembly (–m geno–evalue 0.001–auto-lineage) and annotation (–m prot–evalue 0.001–lineage_dataset ‘metazoa_odb10’) completeness, resulting in 97.6% and 93.1% complete BUSCOs, respectively (Fig. 6). For BUSCO’s annotation completeness, isoforms were filtered from the gene set according to the latest BUSCO protocol78. Finally, we used Merqury73, a reference-free quality and completeness assessment tool for genome assemblies, resulting in 99.54% k-mer completeness and an assembly consensus quality value (QV) of 65.5 (>99.99% accuracy). The final assembly was visualized using Juicebox Assembly Tools35 to identify breakpoints in the assembly. However, we inspected these carefully and found that none show characteristic patterns of read coverage indicative of genuine errors (i.e. misjoins, translocations or inversions).

GenomeScope profile of H. asinina genome. Len: estimated genome length; uniq: overall length of unique, non-repetitive sequences; het: heterozygosity rate; kcov: mean k-mer coverage for first peak; err: error rate; dup: read duplication rate; k: k-mer length (automatically assigned). The figure was generated using GenomeScope 2.074,75.

Code availability
Except where otherwise stated, bioinformatics tools and software were used with default parameters, and all code used for this assembly can be found at https://github.com/roybarkan2020/AbsGenome. In addition, a list of the tools and software used for the assembly is provided in the Methods section (with references to the tool publication, which includes a link to the tool manual and/or GitHub link).
References
OBIS (2022) Distribution records of Haliotis (Linnaeus, 1758). Available: Ocean Biodiversity Information System. Intergovernmental Oceanographic Commission of UNESCO. www.obis.org (2022).
Lee, L. et al. Drawing on indigenous governance and stewardship to build resilient coastal fisheries: People and abalone along Canada’s northwest coast. Mar Policy 109 (2019).
Menzies, C. R. Dm sibilhaa’nm da laxyuubm Gitxaała: Picking Abalone in Gitxaała Territory. Human Organization 69(3), 213–220 (2010).
Field, L. W. et al. Abalone Tales: Collaborative Explorations of Sovereignty and Identity in Native California. (Duke University Press, 2008).
Cook, P. A. The Worldwide Abalone Industry. Modern Economy 5, 1181–1186 (2014).
Hernández-Casas, S. et al. Analysis of supply and demand in the international market of major abalone fisheries and aquaculture production. Mar Policy 148 (2023).
Cook, P. A. & Roy Gordon, H. World abalone supply, markets, and pricing. Journal of Shellfish Research 29, 569–571 (2010).
Vandepeer, M. & Hutchinson, W. G. Abalone Aquaculture Subprogram: Preventing Summer Mortality of Abalone in Aquaculture Systems by Understanding Interactions between Nutrition and Water Temperature. (SARDI Aquatic Sciences, 2006).
IUCN. 2023. The IUCN Red List of Threatened Species. Version 2023-1. https://www.iucnredlist.org (2023).
IUCN. 2022. Human activity devastating marine species from mammals to corals - IUCN Red List. https://www.iucn.org/press-release/202212/human-activity-devastating-marine-species-mammals-corals-iucn-red-list#:~:text=Populations%20of%20dugongs%20%E2%80%93%20large%20herbivorous,Endangered%20due%20to%20accumulated%20pressures (2022).
Hobday, A. J. et al. A hierarchical approach to defining marine heatwaves. Prog Oceanogr 141, 227–238 (2016).
Smith, K. E. et al. Socioeconomic impacts of marine heatwaves: Global issues and opportunities. Science 374 (2021).
Pearce, A. et al. Department of Fisheries & Western Australian Fisheries and Marine Research Laboratories. The ‘Marine Heat Wave’ off Western Australia during the Summer of 2010/11. (Western Australian Fisheries and Marine Research Laboratories, 2011).
Steven, A., Mobsby, D. & Curtotti, R. Australian fisheries and aquaculture statistics 2018. (2020).
Botwright, N. A. et al. Greenlip abalone (Haliotis laevigata) genome and protein analysis provides insights into maturation and spawning. Polish Annals of Medicine 26 (2019).
Orland, C. et al. A Draft Reference Genome Assembly of the Critically Endangered Black Abalone, Haliotis cracherodii. J Hered 113, 665–672 (2022).
Tshilate, T. S., Ishengoma, E. & Rhode, C. A first annotated genome sequence for Haliotis midae with genomic insights into abalone evolution and traits of economic importance. Mar Genomics 70 (2023).
Nam, B. H. et al. Genome sequence of pacific abalone (Haliotis discus hannai): the first draft genome in family Haliotidae. Gigascience 6, 1–8 (2017).
Masonbrink, R. E. et al. An annotated genome for haliotis rufescens (Red Abalone) and resequenced green, pink, pinto, black, and white abalone species. Genome Biol Evol 11, 431–438 (2019).
Gan, H. M. et al. Best foot forward: Nanopore long reads, hybrid meta-assembly, and haplotig purging optimizes the first genome assembly for the southern hemisphere blacklip abalone (haliotis rubra). Front Genet 10 (2019).
Griffiths, J. S. et al. A draft reference genome of the red abalone, Haliotis rufescens, for conservation genomics. J Hered 113, 673–680 (2022).
Lucas, T., Macbeth, M., Degnan, S. M., Knibb, W. & Degnan, B. M. Heritability estimates for growth in the tropical abalone Haliotis asinina using microsatellites to assign parentage. Aquaculture 259, 146–152 (2006).
Jarayabhand, P. & Paphavasit, N. A Review of the Culture of Tropical Abalone with Special Reference to Thailand. Aquaculture 140 (1996).
Mcnarnara, D. C. & Johnson, C. R. Growth of the Ass’s Ear Abalone (Haliotis asinina) on Heron Reef, Tropical Eastern Australia. Mar Freshwater Res 46 (1995).
Maliao, R. J., Webb, E. L. & Jensen, K. R. A survey of stock of the donkey’s ear abalone, Haliotis asinina L. in the Sagay Marine Reserve, Philippines: Evaluating the effectiveness of marine protected area enforcement. Fish Res 66, 343–353 (2004).
Salayo, N. D. et al. Stock enhancement of abalone, Haliotis asinina, in multi-use buffer zone of Sagay Marine Reserve in the Philippines. Aquaculture 523 (2020).
Jarayabhand, P., Yom-La, R. & Popongviwat, A. Karyotypes of marine molluscs in the family Haliotidae found in Thailand. J Shellfish Res 17, 761–764 (1998).
Extracting HMW DNA from Aplysia Tissue Using Nanobind® Kits. https://www.pacb.com/wp-content/uploads/Procedure-checklist-Extracting-HMW-DNA-from-Aplysia-tissue-using-Nanobind-kits.pdf (2022).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods 18, 170–175 (2021).
Daley, T. & Smith, A. Predicting the molecular complexity of sequencing libraries. Nat Methods 10, 325 (2013).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Open2C et al. Pairtools: from sequencing data to chromosome contacts. bioRxiv https://doi.org/10.1101/2023.02.13.528389 (2023).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10 (2021).
Barnett, D. W., Garrison, E. K., Quinlan, A. R., Strömberg, M. P. & Marth, G. T. BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics 27, 1691–1692 (2011).
Durand, N. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst 3, 95–98 (2016).
Zhou, C., McCarthy, S. A. & Durbin, R. YaHS: yet another Hi-C scaffolding tool. Bioinformatics 39, btac808 (2023).
Armin T et al. PacBio Secondary Analysis Tools on Bioconda https://github.com/PacificBiosciences/pbbioconda (2023).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Uliano-Silva, M. et al. MitoHiFi: a python pipeline for mitochondrial genome assembly from PacBio High Fidelity reads. BMC Bioinformatics 24, 288 (2023).
Allio, R. et al. MitoFinder: Efficient automated large-scale extraction of mitogenomic data in target enrichment phylogenomics. Mol Ecol Resour 20, 892–905 (2020).
Laslett, D. & Canbäck, B. ARWEN: a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 24, 172–175 (2008).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. PNAS 117, 9451–9457 (2020).
Smit, A., Hubley, R. & Green, P. RepeatMasker Open-4.0. http://www.repeatmasker.org (2013).
Zhang, Y. et al. Diversity, function and evolution of marine invertebrate genomes. bioRxiv https://doi.org/10.1101/2021.10.31.465852.
Fielman, K. T. & Marsh, A. G. Genome complexity and repetitive DNA in metazoans from extreme marine environments. Gene 362, 98–108 (2005).
Angerer, R. C., Davidson, E. H. & Britten, R. J. DNA Sequence Organization in the Mollusc Aplysia Californica. Cell 6 (1975).
Kamalay, J. C., Ruderman, J. V. & Goldberg, R. B. DNA sequence repetition in the genome of the American oyster. Biochimica et biophysica acta 432(2), 121–128 (1976).
Grüning, B. et al. Bioconda: sustainable and comprehensive software distribution for the life sciences. Nat Methods 15, 475–476 (2018).
Kuo, R. I. et al. Illuminating the dark side of the human transcriptome with long read transcript sequencing. BMC Genomics 21 (2020).
Dainat, J. AGAT: Another Gff Analysis Toolkit to handle annotations in any GTF/GFF format. https://doi.org/10.5281/zenodo.3552717 (2020).
Camacho, C. et al. BLAST+: Architecture and applications. BMC Bioinformatics 10 (2009).
Sayers, E. W. et al. Database resources of the national center for biotechnology information. Nucleic Acids Res 50, D20–D26 (2022).
Consortium, T. U. UniProt: the Universal Protein Knowledgebase in 2023. Nucleic Acids Res 51, D523–D531 (2023).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res 51, D587–D592 (2023).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Science 28, 1947–1951 (2019).
Blum, M. et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res 49, D344–D354 (2021).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res 47, D309–D314 (2019).
NCBI BioProject https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1080039 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR28083764 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR28084366 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR28084367 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR28084368 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR28084369 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR28084370 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR28100643 (2024).
Barkan, R., Strugnell, J., Cooke, I., Watson, S.-A. & Lau, S. Haliotis asinina isolate JCU_RB_2024, whole genome shotgun sequencing project https://identifiers.org/ncbi/insdc:JBANBI000000000.1 (2024).
Barkan, R. Annotation files for Haliotis asinina genome assembly. Figshare https://doi.org/10.6084/m9.figshare.25283317.v3 (2024).
Barkan, R. Repeat sequences analysis files for Haliotis asinina genome assembly. Figshare https://doi.org/10.6084/m9.figshare.25284904.v1 (2024).
Barkan, R. Mitochondrial genome assembly files for Haliotis asinina genome assembly. Figshare https://doi.org/10.6084/m9.figshare.25283329.v1 (2024).
Barkan, R. Genome methylation regions file for Haliotis asinina genome. Figshare https://doi.org/10.6084/m9.figshare.26501332.v1 (2024).
Sim, S. B., Corpuz, R. L., Simmonds, T. J. & Geib, S. M. HiFiAdapterFilt, a memory efficient read processing pipeline, prevents occurrence of adapter sequence in PacBio HiFi reads and their negative impacts on genome assembly. BMC Genomics 23 (2022).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: Reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol 21 (2020).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat Commun 11 (2020).
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075 (2013).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Manni, M., Berkeley, M. R., Seppey, M. & Zdobnov, E. M. BUSCO: Assessing Genomic Data Quality and Beyond. Curr Protoc 1 (2021).
Hao, Z. et al. RIdeogram: drawing SVG graphics to visualize and map genome-wide data on the idiograms. PeerJ Comput Sci 6, e251 (2020).
Chen, C. et al. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol Plant 13, 1194–1202 (2020).
Acknowledgements
The authors would like to acknowledge the Marine and Aquaculture Research Facility (MARF, James Cook University, Townsville) team, Cairns Marine (Cairns, Queensland, Australia) for collecting the abalone, the Australian Genome Research Facility (AGRF) team – Dr Dhanya Sooraj, Trent Peters and Saurabh Shrivastava. We would also like to acknowledge Dr Inga A. Frøland Steindal, Dr Bruna Louise Pereira Luz and Julia Yun-Hsuan Hung for their lab support.
Author information
Authors and Affiliations
Contributions
Study design: R.B., J.S., I.C. and S.A.W. Laboratory work: R.B. and S.C.Y.L. Data analysis and interpretation: R.B., J.S., I.C. and S.C.Y.L. Drafting the manuscript: R.B., J.S., I.C., S.A.W. and S.C.Y.L.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Barkan, R., Cooke, I., Watson, SA. et al. Chromosome-scale genome assembly of the tropical abalone (Haliotis asinina). Sci Data 11, 999 (2024). https://doi.org/10.1038/s41597-024-03840-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03840-w
