

Abstract
T-type calcium channels play critical roles in cellular excitability and have been implicated in the pathogenesis of a variety of neurological disorders including epilepsy. Although there have been reports that certain neuroleptics that primarily target D2dopamine receptors and are used to treat psychoses may also interact with T-type Ca channels, there has been no systematic examination of this phenomenon. In the present paper we provide a detailed analysis of the effects of several widely used neuroleptic agents on a family of exogenously expressed neuronal T-type Ca channels (α1G, α1H, and α1Isubtypes). Among the neuroleptics tested, the diphenylbutylpiperidines pimozide and penfluridol were the most potent T-type channel blockers with Kd values (∼30–50 nm and ∼70–100 nm, respectively), in the range of their antagonism of the D2 dopamine receptor. In contrast, the butyrophenone haloperidol was ∼12- to 20-fold less potent at blocking the various T-type Ca channels. The diphenyldiperazine flunarizine was also less potent compared with the diphenylbutylpiperadines and preferentially blocked α1G and α1I T-type channels compared with α1H. The various neuroleptics did not significantly affect T-type channel activation or kinetic properties, although they shifted steady-state inactivation profiles to more negative values, indicating that these agents preferentially bind to channel inactivated states. Overall, our findings indicate that T-type Ca channels are potently blocked by a subset of neuroleptic agents and suggest that the action of these drugs on T-type Ca channels may significantly contribute to their therapeutic efficacy.
The rapid entry of calcium into cells through voltage-dependent Ca channels triggers a variety of intracellular events such as muscle contraction, hormone secretion, synaptic transmission, and gene expression. Ca channels have been traditionally classified into high voltage-activated (HVA) and low voltage-activated (LVA) subtypes (Tsien et al., 1988; Bean, 1989). HVA Ca channels first activate at relatively depolarized potentials and comprise L-, P/Q-, N-, and R-types. These channels exhibit a high single-channel conductance and varied patterns of inactivation and deactivation. LVA Ca channels, also known as T-type, show a more negative range of activation and inactivation, rapid inactivation, slow deactivation, and smaller single-channel conductances. T-type Ca channels are present in a variety of cell types where they appear to mediate low-threshold spikes and rebound burst firing patterns (for review, see Huguenard, 1996), pacemaker activity (Hagiwara et al., 1988), hormone secretion (Rossier et al., 1996), cell growth and proliferation (Xu and Best, 1990), and are also involved in fertilization (Arnoult et al., 1996; Santi et al., 1996). Abnormal activity of T-type Ca channels has been implicated in the pathophysiology of epilepsy (Huguenard and Prince, 1994; Tsakiridou et al., 1995; Huguenard, 1996), hypertension (Self et al., 1994), and cardiac hypertrophy (Nuss and Houser, 1993). Furthermore, the activity of T-type channels may play a role in enlarging the area of damage produced by a transient ischemic insult (Ito et al., 1994).
Three different T-type Ca channels have been cloned and expressed from mammals: α1G, α1H, and α1I (Cribbs et al., 1998; Perez-Reyes et al., 1998; Klugbauer et al., 1999; Lee et al., 1999; McRory et al., 2001). Although HVA Ca channels have a well defined pharmacology, it has proven difficult to identify specific high-affinity blockers of LVA Ca channels. Ni2+ and amiloride are often described as effective blockers of T-type channels in many native cells, although the effects of these agents is quite variable in different cell types (Huguenard, 1996; Todorovic and Lingle, 1998).
Neuroleptic agents comprise several chemically distinct classes of compounds (diphenylbutylpiperidines, butyrophenones, and phenothiazine) that act as antagonists of the D2 dopamine receptors and are widely prescribed to treat a variety of psychiatric disorders (Seeman et al., 1976). Interestingly, many of these drugs also display Ca channel blocking activity, although their selectivity for the different Ca channel subtypes has not been systematically examined. Although some neuroleptics inhibit to some extent P-, N-, and L-type Ca channels, they appear to block T-type channels with relatively higher affinity than HVA Ca channels (Enyeart et al., 1990,1993; Takahashi and Akaike, 1991; Sah and Bean, 1993). The diphenylbutylpiperidines (DPBPs) pimozide and penfluridol have been shown to block T-type currents in various cell types: heart (Enyeart et al., 1990), adrenal fasciculata (Enyeart et al., 1993), mouse spermatogenic (Arnoult et al., 1998), neural crest cells, and human C cell lines (Enyeart et al., 1992). Moreover, the neuroleptic-like agent flunarizine has also been reported to act both as a D2 dopamine receptor antagonist and as blocker of Ca currents in the CNS where it acts as a potent organic blocker of the T-type currents from dissociated hypothalamic neurons (Akaike et al., 1989) as well as hypothalamic T-type currents expressed inXenopus oocytes (Dzura et al., 1996).
In the present paper we explored the inhibitory effects of the neuroleptic agents pimozide, penfluridol, haloperidol, and flunarizine on three exogenously expressed neuronal T-type Ca channels. The results demonstrate that the DPBP class of neuroleptics are high-affinity T-type Ca channel blockers and suggest that Ca channel block may represent a significant contribution to the clinical efficacy of these agents.
MATERIALS AND METHODS
Cell culture. Stable cell lines expressing α1G, α1H, or α1I were generated by transfecting either α1G, α1H, or α1I cDNAs (all in pcDNA3.1 vector) into human embryonic kidney (HEK) tsa 201 cells using standard Ca-phosphate precipitation, and clones were selected with zeocin. In some cases standard Ca-phosphate precipitation was also used for transient cotransfection in HEK tsa 201 cells of α1I (3 μg in pcDNA3.1 vector), CD8 (2 μg) marker plasmid, and 15 μg of pBluescript SK carrier DNA for a total of 20 μg of cDNA mix. Transiently transfected cells were selected for expression of CD8 by adherence of Dynabeads (Dynal, Great Neck, NY). The cells were grown in standard DMEM (10% fetal bovine serum and 50 U/ml penicillin–streptomycin), maintained at 37°C in a humidified atmosphere of 95% O2 and 5% CO2. Stably expressing cell lines were enzymatically dissociated with trypsin–EDTA and plated on 35 mm Petri dishes 12 hr before recordings. Functional transient expression of α1I was evaluated 24 hr after transfection.
Electrophysiological recordings. Macroscopic currents were recorded using the whole-cell patch-clamp technique (Hamill et al., 1981). The external recording solution contained in mm: 2 CaCl2 or 2 BaCl2, 1 MgCl2, 10 HEPES, 40 TEA Cl, 92 CsCl, and 10 glucose, pH 7.2. The internal pipette solution contained in mm: 105 CsCl, 25 TEA Cl, 1 CaCl2, 11 EGTA, and 10 HEPES, pH 7.2. α1G and α1I currents were recorded in the presence of 2 mm external Ca. Because α1H Ca currents are significantly smaller than Ba currents (McRory et al., 2001), we used Ba as a charge carrier for α1H (Ca was also used where indicated). Whole-cell currents were recorded using an Axopatch 200B or 200A amplifier (Axon Instruments, Foster City, CA), controlled and monitored with a personal computer running pClamp software version 6.03 (Axon Instruments). Patch pipettes (borosilicate glass BF150-86-10; Sutter Instruments, Novato, CA), were pulled using a Sutter P-87 puller and fire-polished using a Narishige (Tokyo, Japan) microforge, with typical resistances of 2.5–4 MΩ (filled with internal solution). Series resistance was electronically compensated by at least 60%. Whole-cell currents that exceeded 2 nA were not examined, minimizing voltage error (<2–3 mV). Only cells exhibiting adequate voltage control [judged by smoothly rising current–voltage (I–V) relationship and monoexponential decay of capacitive currents] were included in the analysis. The bath was connected to ground via a 3 m KCl Agar bridge. All recordings were performed at room temperature (20–24°C).
Drugs effects were investigated using 150 msec steps to peak potentials every 15–30 sec from a holding potential of −100 mV. Current–voltage relations were measured by a series of 150-msec-long depolarizing pulses applied from a holding potential of −100 mV to membrane potential between −90 and +20 mV every 15 sec.
Data were low-pass filtered at 2 kHz using the built-in Bessel filter of the amplifier, and in most cases, subtraction of capacitance and leakage current was performed on-line using P/4 protocol. Recordings were analyzed using Clampfit 6.03 (Axon Instruments), and figures were generated using the software program Microcal Origin (version 3.78). Data from drug concentration–response studies were fitted with the equation y = [(A1−A2)/{1 + (x/xo)P} + A2, whereA1 is initial (= 0) andA2 final value,xo is IC50(concentration causing 50% inhibition of currents), and pgives a measure of steepness of curve.
Time courses of channel blockade were well described by single exponential curves from which τon and the fraction of unblocked channels at equilibrium (a =Idrug/Icontrol) were obtained. Kd values were estimated using the following equation:
 The steady-state inactivation curves were constructed by plotting the normalized current during the test pulse as a function of the conditioning potential. The data were fitted with a Boltzmann equation: I/Imax = {1 + exp[V − V0.5i/ki]}−1, where I is the peak current,Imax is the peak current when the conditioning pulse was −120 mV, V andV0.5i are the conditioning potential and the half-inactivation potential respectively, andki is the inactivation slope factor.
The steady-state inactivation curves were constructed by plotting the normalized current during the test pulse as a function of the conditioning potential. The data were fitted with a Boltzmann equation: I/Imax = {1 + exp[V − V0.5i/ki]}−1, where I is the peak current,Imax is the peak current when the conditioning pulse was −120 mV, V andV0.5i are the conditioning potential and the half-inactivation potential respectively, andki is the inactivation slope factor.
Current–voltage relationships were fitted with the Boltzmann equation:I = {1/1 + exp(Vm −V0.5a)/ka)}{(V−Erev)Gmax}, where Vm is the test potential,V0.5a is the half-activation potential, Erev is the extrapolated reversal potential, Gmax is the maximum slope conductance, and kareflects the slope of the activation curve.
Statistical significance was determined by Student's t test or one-way ANOVA followed by Bonferroni multiple comparison post-test, as appropriate, and significant values were set as indicated in the text and figure legends.
Solutions and drugs. Penfluridol was from Research Diagnostics Inc. (Flanders, NJ). All other drugs were purchased from Sigma (St. Louis, MO), unless otherwise specified. Test solutions containing drugs were prepared fresh for each experiment from concentrated stock solutions and added to the recording solution. Concentrated stock solutions were as follows: pimozide (1 mm in DMSO); penfluridol (500 μm in ethanol), haloperidol (1 mm in DMSO), and flunarizine (1 mm in DMSO). The highest concentration of ethanol and DMSO in the recording solution did not exceed 0.1%, a concentration that did not detectably affect calcium channel properties. The perfusion system consisted of a custom-made multiple solution perfusion manifold consisting of three input and three output capillary tubes (custom microfil, 28 gauge, 250 μm inner diameter and 350 μm outer diameter; World Precision Instruments, Sarasota, FL) ensheathed in a glass pipette. The lengths of lines from valve to the manifold were kept to a minimum, and the outputs of the manifold were placed within five cell lengths, resulting in cells being bathed with new solutions with minimal delay (within 1 sec) and minimal dead space volume.
RESULTS
Diphenylbutylpiperidines are high-affinity antagonists of T-type Ca channels
The DPBPs (e.g., pimozide, penfluridol) (Fig.1A,B) are a class of neuroleptic drugs clinically used to relieve the symptoms of schizophrenia (Pinder et al., 1976). The DPBPs were initially identified as potential Ca channel antagonists when it was discovered that these agents inhibited dihydropyridine (DHP) binding to specific brain receptors and also inhibited depolarization-dependent Ca uptake and contraction in muscle (Gould et al., 1983). A number of reports have shown that DPBPs are relatively potent blockers of L-type Ca channels in endocrine (e.g., pituitary) and heart muscle cells (Enyeart et al., 1990). In some of these same preparations it has been observed that low concentrations of DPBPs also block T-type currents (Enyeart et al., 1990, 1992). While some DPBPs (e.g., pimozide) have been used to probe the physiological functions of T-type channels (Arnoult et al., 1998), it has not been established whether these agents are selective for particular subtypes of T-type channel. We tested the effect of two DPBPs, penfluridol and pimozide (Fig. 1A,B), on exogenously expressed α1G, α1H, and α1I T-type Ca channels from rat brain (Fig. 2). Penfluridol and pimozide were not subtype-selective because both of these agents blocked the three neuronal T-type Ca channels to similar extents (Fig. 2, Table 1). A detailed analysis of the concentration–response relations of pimozide and penfluridol with the three T-type Ca channels revealed a similar range of IC50 values in the case of pimozide (34.6, 53.5, and 30.4 nm for α1G, α1H, and α1I, respectively) and IC50 values equal to 93.1, 64.1, and 71.6 nm for α1G, α1H, and α1I, respectively, for penfluridol (Fig. 2A,B)

Chemical structures of the three different structural classes of neuroleptic agents used in this study: the DPBPs pimozide (A) and penfluridol (B). An example of a butyrophenone (C, haloperidol) and a diphenyldiperazine, flunarizine (D).

Summary of the effect of DPBPs on T-type calcium channels. A, B, Concentration–response curves for pimozide and penfluridol of α1G, α1H, and α1I T-type channels. Data points reflect mean ± SE of two to six determinations. Half-maximal inhibition (IC50) were determined from fitting the data as described in the methods. IC50 values for pimozide were 34.6, 53.5, and 30.4 nm, for α1G, α1H, and α1I, respectively. IC50 values for penfluridol were 93.1, 64.1, and 71.6 nm, for α1G, α1H, and α1I, respectively. Inset, Representative current traces in control (filled circles) and 5 min after addition of drugs (open circles). C, D, Time course of block of α1G currents in eight different cells exposed to 10, 50, 500, and 1000 nmpimozide and 50, 200, 1000, and 10,000 nm penfluridol. Ca currents were evoked every 15 sec with 150 msec step depolarizations from −100 to −40 mV. Solid lines, Fits of single exponential decays to a plateau. Insets, Dependence of 1/τon on [drug]. Values are mean and SE of three to five determinations. Solid line fit was made with the following equation: 1/τon =kon[drug] +koff, wherekon = 0.00432/sec,koff = 0.000088/sec, andKd = 48.4 nm for pimozide, and kon = 0.00117/sec,koff = 0.0000087/sec, andKd = 135.1 nm for penfluridol.
Differential affinity of three classes of neuroleptic agents for three cloned T-type channels
Examination of current–voltage relations and kinetic parameters showed that the DPBPs did not significantly affect these properties of T-type Ca channels (data not shown).
The blocking rate of penfluridol was ∼10 times slower than pimozide with τon values at 1 μm of 98.5 ± 17.2 sec (n = 5) for penfluridol and 10.01 ± 1.8 sec (n = 3) for pimozide (Fig.2C,D, see for the α1G data). The blocking effect of both drugs was faster at higher concentrations. The time course of onset for pimozide and penfluridol block for α1G could be fitted well with a single exponential (Fig. 2C,D), and the plot of 1/τon exhibited a linear concentration dependence as would be expected for a 1:1 interaction (Fig. 2C,D, insets). The values of the binding (kon) and unbinding (koff) rate constants were obtained from the slope and y-intercept, respectively, of the linear equation with Kd values of 48.8 and 135.1 nm for pimozide and penfluridol, respectively, which are very close to the IC50values obtained from the dose–response curves. Washout of pimozide was faster and more complete than for penfluridol (see Fig.5A,B), and washout of penfluridol was largely incomplete. The extent of recovery could be increased only slightly by holding at more negative potentials (data not shown). Complete recovery of DPBP block has been seen for T-type currents in some native cells (Enyeart et al., 1992), whereas in other cell types DPBP block is reported to be more persistent (Arnoult et al., 1998).
The butyrophenone haloperidol blocks T-type channels with lower affinity than DPBPs
Haloperidol (Fig. 1C) is a potent butyrophenone antipsychotic commonly used to treat disorders such as schizophrenia (DiMascio, 1972). Cardiac arrhythmias have been associated with haloperidol therapy (Hunt and Stern, 1995), although the mechanism for this effect is unclear. It has been proposed that haloperidol could mediate cardiac effects by the block of repolarizing potassium currents, in particular HERG channels (Hunt and Stern, 1995). However, because T-type Ca channels are present in pacemaker heart cells, they may also be involved in the pathogenesis of arrhythmias. Figure 3 summarizes the effects of application of 1 μm haloperidol on α1G, α1H, and α1I T-type Ca channels. The estimatedKd values for haloperidol interaction were similar among all three T-type channel subtypes (range, 1.2–1.4 μm) (Table 1). However, haloperidol is 12-to 20-fold less potent on the T-type channels compared with the DPBP neuroleptics pimozide and penfluridol (Table 1). The block of haloperidol developed faster than the other neuroleptic drugs with τon values ranging from 13 to 20 sec, and drug washout was similarly faster and more complete (see Fig.5C).
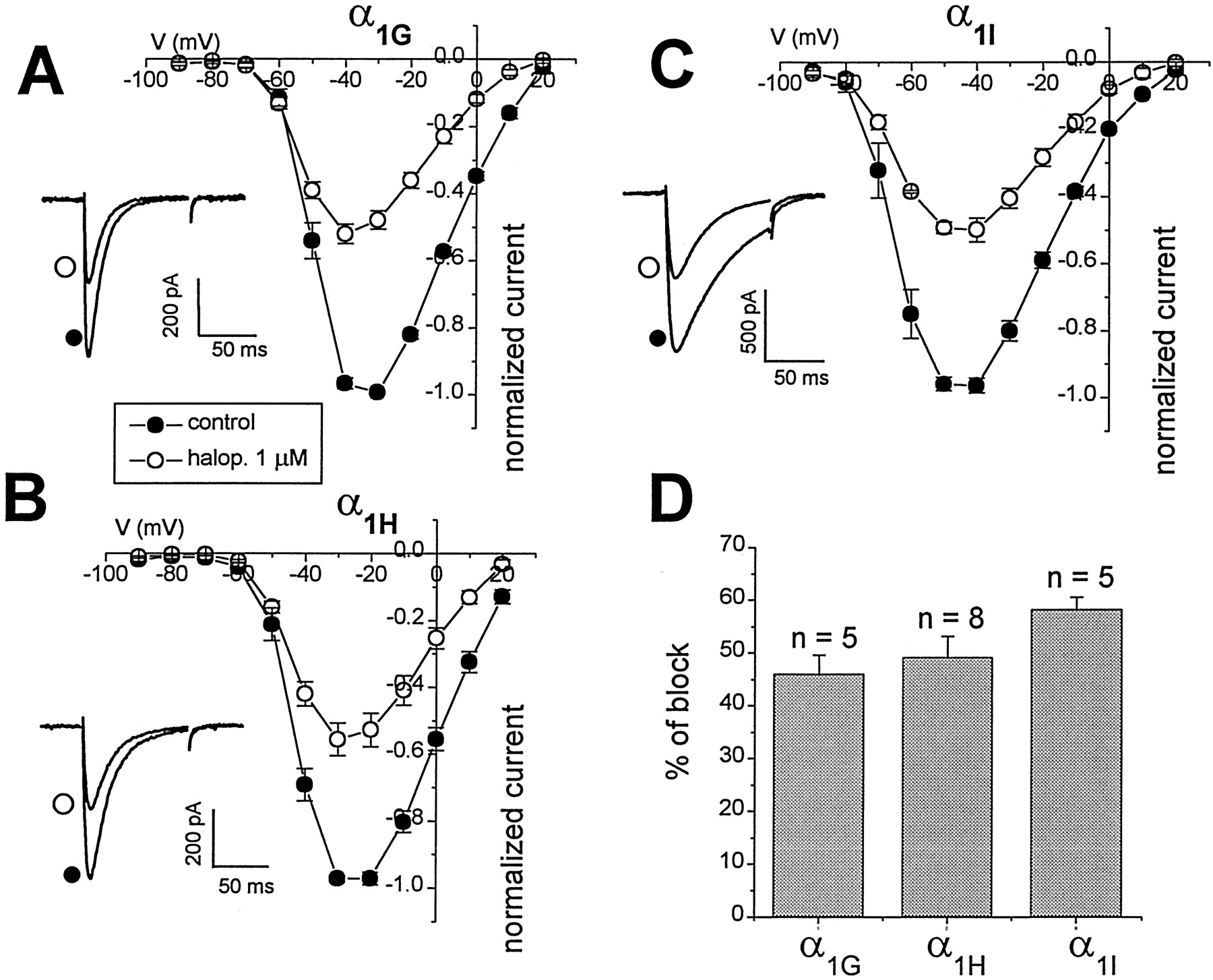
Summary of the effect of the butyrophenone haloperidol, on α1G, α1H, and α1I T-type Ca channels. Mean I–Vrelationships of T-type currents in the absence (filled circles) or presence (open circles) of haloperidol (1 μm) on currents through α1G(A), α1H (B), and α1I (C). Normalized meanI–V data represent the mean ± SE from three to eight cells. Insets show representative current traces in absence (control, filled circles) and presence (open circles) of 1 μm haloperidol, elicited with test pulses to −40 mV (α1G, α1I) or −30 mV (α1H).D, All currents were inhibited to similar levels (≈50% inhibition) by 1 μm haloperidol, as summarized in this bar chart (n = 5 for α1G; n = 8 for α1H; n = 5 for α1I).
Flunarizine exhibits subtype-specific blocking activity for T-type calcium channels
Flunarizine is a diphenyldiperazine derivative (Fig.1D) that has been used clinically to treat a number of diseases such as vertigo (Olesen, 1988), migraine (Spierings, 1988), and some heart disorders (Koch et al., 1990). Flunarizine is also a potent anticonvulsant (Greenberg, 1987) and displays some neuroprotective properties (Desphande and Wieloch, 1986; Rich and Hollowell, 1990; Kaminski Schierle et al., 1999). Flunarizine has been shown to block native L-, N-, and T-type Ca channels (Tytgat et al., 1988, 1991, 1996), and some neurons possess T-type currents characterized by their high sensitivity to this neuroleptic compound (Kaneda and Akaike, 1989; Panchenko et al., 1993; Takahashi and Akaike, 1991). In certain preparations, flunarizine has been described as the most potent organic blocker of hypothalamic T-type Ca channels (Akaike et al., 1989; Dzura et al., 1996).
Figure 4 summarizes the effects of 1 μm flunarizine on α1G, α1H, and α1I T-type Ca channels. Unlike the other neuroleptics studied, flunarizine displayed a preferential block of α1G and α1I (Kd = 0.53 and 0.84 μm, respectively) (Table 1) compared with α1H (Kd = 3.6 μm) (Table 1). As with pimozide, penfluridol, and haloperidol, flunarizine did not significantly alter the current–voltage relations or current kinetics of the three T-type channels (Fig. 4). The onset of blockade by flunarizine was the slowest (Fig. 5D) of the neuroleptics tested with τon values at 1 μm ranging from 72 to 197 sec for α1G and α1I. Washout of flunarizine was likewise slow and essentially complete.

Differential blockade of T-type Ca channels by the diphenyldiperazine flunarizine. Mean I–V relationships of T-type currents in the absence (filled circles) or presence (open circles) of flunarizine (1 μm), were obtained from normalized currents (mean ± SE from 3–6 cells) through α1G(A), α1H (B), and α1I (C). Normalized meanI–V data represent the mean ± SE from three to six cells. Insets show representative current traces in absence (control, filled circles) and presence (open circles) of 1 μm flunarizine, elicited with test pulses to −40 mV (α1G, α1I) or −30 mV (α1H).D, Flunarizine (1 μm) differentially inhibited the cloned T-type currents, inhibiting α1G and α1I more potently (∼70%) than α1H(∼30%).

Time course of neuroleptic blockade of the three T-type Ca channels. Shown are representative time courses of the inhibitory effects of 500 nm pimozide (A), 1 μm penfluridol (B), 1 μm haloperidol (C), and 1 μm flunarizine (D) on cloned rat α1G, α1H, and α1I channels. Drug application (first arrow) followed by washout of drugs (second arrow) appear as indicated. The DPBPs penfluridol (1 μm) and pimozide (500 nm), showed τon of inhibition ranging from 60.3 ± 16 to 109.9 ± 29 and from 29.5 ± 0.4 to 84.2 ± 12.4 sec (n = 3–6) for penfluridol and pimozide, respectively. In contrast, the butyrophenone haloperidol showed τon values from 13.3 ± 0.9 to 20 ± 2.6 sec (n = 4). Flunarizine exhibits the slowest on-rate (from 39.2 ± 8.8 to 197.3 ± 23.8 sec; n= 3). The T-type Ca currents were elicited by test pulses to −40 mV (α1G, α1I) or −30 mV (α1H) from −100 mV holding potential (every 15 sec before and during drug application).
Neuroleptics affect T-type Ca channel steady-state inactivation properties
To further characterize the biophysical effects of neuroleptics on the three types of neuronal T-type Ca channels, a detailed analysis of voltage-dependent inactivation of the α1G Ca channel was performed. A conditioning pulse of 15 sec duration at various potentials between −120 and −50 mV was applied and followed by a test pulse to −30 mV of 150 msec duration. Figure6 shows that addition of flunarizine, haloperidol, pimozide, and penfluridol caused a significant hyperpolarizing shift (from 7 to 10 mV) ofV0.5inact compared with the steady-state inactivation profiles of the α1Gcurrents under control conditions. Similar voltage-dependent block of α1H and α1I channels by penfluridol, haloperidol, or flunarizine is suggested by comparing data at more depolarized potentials (i.e., −85 vs −100 mV; data not shown).

Neuroleptic agents induce a negative shift in the voltage dependence of steady-state inactivation of α1GT-type Ca channels. The V0.5i values of rat α1G channel inactivation were shifted to hyperpolarized potentials as follows: A, 50 nm pimozide (control V0.5i = −78.6 ± 0.2 mV vs treated V0.5i = −85.8 ± 0.2 mV; p < 0.01; n = 4).B, 100 nm penfluridol (controlV0.5i = −78.5 ± 0.3 mV vs treated V0.5i = −90.2 ± 0.3 mV;p < 0.01; n = 4).C, 1 μm haloperidol (controlV0.5i = −80.8 ± 0.2 mV vs treated V0.5i = −91.1 ± 0.3 mV;p < 0.01; n = 3).D, 100 nm flunarizine (controlV0.5i = −81.6 ± 0.3 mV vs treated V0.5i = −90.4 ± 0.2 mV;p < 0.01; n = 4). Steady-state inactivation curves were generated as described in Materials and Methods.
Whereas inactivation-gating processes were profoundly affected by all neuroleptic drugs tested, the voltage dependence of activation was not significantly altered. The V0.5 of activation obtained from I–V curves were not significantly shifted (<5 mV) in the presence of the drugs (data not shown).
DISCUSSION
Neuroleptics represent chemically diverse classes of compounds that share the ability to alleviate the symptoms of CNS disorders such as schizophrenia and some mood disorders. Several previous studies have shown that these agents can inhibit native T-type Ca channels in a number of cell types (Enyeart et al., 1990, 1992; Arnoult et al., 1998). In a systematic analysis of the cloned α1G, α1H, and α1I T-type Ca channel subtypes, we show here that distinct classes of neuroleptics: the DPBPs (pimozide and penfluridol), butyrophenones (haloperidol), and the diphenyldiperazine derivative flunarizine, differentially affect neuronal T-type Ca channels.
Diphenybutylpiperidines are potent blockers of T-type Ca channels
The DPBPs are a class of neuroleptic agents used in the treatment of schizophrenia and related psychoses such as delusional disorder. Pimozide is also used in the treatment of Tourette's syndrome, which is a familial neurobehavioral disorder characterized by fluctuating involuntary motor and/or vocal tics (Cohen et al., 1992). It is thought that DPBPs owe much of their antipsychotic effectiveness to their antagonism of dopamine receptors (Seeman et al., 1976). However, several studies have shown they also interact with other nervous system targets, including Ca channels (Enyeart et al., 1992; Sah and Bean, 1993). Our results have identified the DPBPs penfluridol and pimozide as two of the most effective T-type Ca channel antagonists among the available organic blockers. We find that theKd values for pimozide (39–60 nm) are very similar to publishedKd values for dopamine D2 receptors (29 nm;Richelson and Souder, 2000). Studies on neural crest-derived cell lines and adrenal zona fasciculata (AZF) cells have shown that penfluridol potently blocks T-type channels with IC50 values of 224 nm (Enyeart et al., 1992) and 300 nm (Enyeart et al., 1993), respectively, whereas pimozide inhibited T-type currents in the AZF cells and spermatogenic cells with IC50 of 500 and 460 nm, respectively. Because the concentration of permeant ion has been shown to affect piperidine blocking affinity (Zamponi et al., 1996), the discrepancy between the blocking potency observed in native T-type channels and that which we report in the cloned channels may be explained by the different experimental conditions used (10 mm Ca was used in native channels vs 2 mm Ca/Ba used in our experiments). To explore this possibility, experiments were performed in 10 mm external Ca for comparison. These experiments showed that 1 μm penfluridol blocked α1G 71.6 ± 5.6% (n = 3) in 10 mm Ca, whereas the same concentration blocked T-type currents 90.9 ± 2.8 (n = 7) α1G in 2 mm Ca (data not shown). Thus, the difference in Ca concentrations probably accounts for only a small portion of the difference.
The potency of DPBPs as T-type Ca channel antagonists parallels their potency as D2 receptor antagonists (cf. Gould et al., 1983; this study). In contrast, butyrophenone and phenothiazine are much weaker Ca channel antagonists compared with D2 dopamine receptor blockers (haloperidolKd values: D2receptor, 2.6 nm, Richelson and Souder, 2000; T-type channels, ∼1200 nm; this study). Thus, it is possible that at therapeutic doses DPBPs occupy receptor sites on both T-type Ca channels and D2 dopamine receptors, whereas butyrophenones like haloperidol bind almost exclusively to D2 dopamine receptors. Clinically, DPBPs offer an advantage over other drugs (e.g., butyrophenones) because they are able to relieve the negative symptoms of schizophrenia such as emotional withdrawal and poverty of speech that are resistant to treatment with other classical neuroleptic drugs. The only known pharmacological action that distinguishes DPBPs from other neuroleptics such as the butyrophenones (e.g., haloperidol) is their high-potency T-type Ca-antagonist activity. This raises the possibility that the selective clinical improvement in negative symptoms observed for DPBPs is attributable to their ability to block T-type calcium channels (Gould et al., 1983; Snyder and Reynolds, 1985).
Neuroleptic agents preferentially bind to the inactivated state
We find that the effects of all the neuroleptic drugs examined are potentiated at more depolarized holding potentials. This suggests that these drugs bind with higher affinity to the inactivated state of the channels. Similar results in native cells showed that penfluridol (300 nm) and pimozide (500 nm) shifted the steady-state inactivation of T-type currents by −8.2 and −10 mV, respectively (Enyeart et al., 1993). In addition T-type currents from spermatogenic cells have been shown to be blocked by pimozide in a voltage-dependent manner (Arnoult et al., 1998). Flunarizine has also been reported to have voltage-dependent inhibitory effects on many native T-type currents; for example, 5 μm flunarizine shifted the steady-state inactivation of T-type currents in rat Purkinje neurons by −10 mV (Panchenko et al., 1993) and similarly in mouse neuroblastoma cells (Wang et al., 1990) and guinea pig ventricular myocytes (Tytgat et al., 1996). This voltage dependence of block is particularly relevant in cells that fire action potentials because more depolarized potentials would be predicted to enhance the potency of blockade.
Flunarizine preferentially blocks α1G and α1I T-type channels
Flunarizine is a diphenyldiperazine derivative described as a Ca channel antagonist (Tytgat et al., 1988; Akaike et al., 1989; Wang et al., 1990), although it also displays moderate dopamine receptor antagonist activity with reported Kdvalues of 100–500 nm (Ambrosio and Stefanini, 1991; Brucke et al., 1995). Interestingly, these values are similar to those found in our study for α1G and α1I T-type Ca channel blockade (∼530 and 840 nm, respectively) (Table 1). Clinically, flunarizine has been mainly prescribed to treat vertigo (Olesen, 1988), migraine (Spierings, 1988), and epilepsy (Greenberg, 1987). The mechanisms underlying the clinical efficacy of flunarizine are considered to be related to modulation of the activity of neuronal Ca channels (Moron et al., 1989). Flunarizine has been identified as a moderately potent T-type Ca channel blocker in native cells such as hypothalamic neurons (IC50 = 0.7 μm; Akaike et al., 1989). Similar effects of flunarizine on T-type Ca currents have been observed in rat amygdaloid neurons (Kaneda and Akaike, 1989), hippocampal CA1 pyramidal neurons (Takahashi and Akaike, 1991), N1E-115 neuroblastoma cells (Wang et al., 1990), and in Purkinje cells (Panchenko et al., 1993). In contrast, T-type channels from guinea pig ventricular myocytes and rat calcitonin secreting C-cells display lower affinity for this compound (Kd, ∼10 μmand IC50 ∼10 μm,respectively) (Enyeart et al., 1992; Tytgat et al., 1996). The different blocking potencies of flunarizine on native T-type currents could be explained by the recently described diversity in the molecular composition of the T-type channels. Of particular relevance, we report here that among the neuroleptic drugs tested, flunarizine displayed preferential blockade of the α1G and α1I subtypes withKd values approximately five times higher for the α1H channels (Table 1). A similar difference in Kd was observed when Ca was used as a charge carrier for α1H(Kd, 3.04 μm).
The blockade of T-type Ca channels by flunarizine is consistent with its depressive action on repetitive neuronal firing and may contribute to its efficacy in suppressing seizure discharge (Bingmann and Speckmann, 1989). The α1G T-type channel is highly expressed in the thalamus (McRory et al., 2001) and may represent the molecular target for flunarizine action.
In summary, we have characterized the effects of clinically important neuroleptic agents on the α1G, α1H, and α1I T-type Ca channels expressed in the mammalian CNS. The results show that the DPBPs antagonize T-type Ca channels with a potency similar to their affinity for D2 dopamine receptors, and thus Ca channel block may represent a significant contribution to the clinical efficacy of these agents. Flunarizine exhibited preferential block of α1G and α1I compared with the α1H T-type Ca channel that may contribute to the differential therapeutic efficacy of this agent compared with DPBPs. Overall, the inhibitory effects of the DPBP neuroleptics on T-type Ca channels may underlie some of the clinical efficacy (and side effects) of antipsychotic treatments.
Footnotes
This work was supported by a grant from Canadian Institutes for Health Research (CIHR) (T.P.S.), fellowship support from the Human Frontiers Science Program (C.M.S.), from the CIHR (J.E.M.), and from the Natural Sciences and Engineering Research Council of Canada and Killam Foundation (K.S.C.H.), and a CIHR Senior Scientist Award (T.P.S.).
Correspondence should be addressed to Dr. Terrance P. Snutch, Biotechnology Laboratory, Room 237–6174, University Boulevard, University of British Columbia, Vancouver, British Columbia, Canada V6T 1Z3. E-mail: snutch{at}zoology.ubc.ca.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}