

Abstract
Low voltage-activated Ca2+ channels play important roles in pacing neuronal firing and producing network oscillations, such as those that occur during sleep and epilepsy. Here we describe the cloning and expression of the third member of the T-type family, α1I or CavT.3, from rat brain. Northern analysis indicated that it is predominantly expressed in brain. Expression of the cloned channel in either Xenopusoocytes or stably transfected human embryonic kidney-293 cells revealed novel gating properties. We compared these electrophysiological properties to those of the cloned T-type channels α1G and α1H and to the high voltage-activated channels formed by α1Eβ3. The α1I channels opened after small depolarizations of the membrane similar to α1G and α1H but at more depolarized potentials. The kinetics of activation and inactivation were dramatically slower, which allows the channel to act as a Ca2+ injector. In oocytes, the kinetics were even slower, suggesting that components of the expression system modulate its gating properties. Steady-state inactivation occurred at higher potentials than any of the other T channels, endowing the channel with a substantial window current. The α1I channel could still be classified as T-type by virtue of its criss-crossing kinetics, its slow deactivation (tail current), and its small (11 pS) conductance in 110 mm Ba2+ solutions. Based on its brain distribution and novel gating properties, we suggest that α1I plays important roles in determining the electroresponsiveness of neurons, and hence, may be a novel drug target.
Voltage-gated calcium channels can be subdivided into two classes based on the voltage required to trigger channel opening. Low voltage-activated (LVA) Ca2+channels begin to open after small depolarizations (10 mV) of the plasma membrane, whereas high voltage-activated (HVA) channels require much stronger depolarizations (40 mV). Most voltage-gated Na+ channels open somewhere between these two extremes. Entry of Ca2+ ions causes membrane depolarization. LVA Ca2+ channels activate at potentials low enough to gate the activity of other depolarizing voltage-activated ion channels. This led to the hypothesis that LVA channels could act as pacemaker currents, controlling the activity of other voltage-gated ion channels. A clear example of this phenomenon is the thalamic low-threshold Ca2+ spike that is crowned with a burst of action potentials mediated by Na+ channels (Llinas and Jahnsen, 1982). Patch-clamp recordings demonstrated that T-type Ca2+ channels mediated the low-threshold spike and that they are involved in rebound burst firing, oscillations, and resonance (for review, see Huguenard, 1996).
Molecular cloning of ion channels has revealed a greater diversity than was expected from electrophysiological studies of endogenous currents. For HVA Ca2+ channels, there are at least seven genes encoding α1 subunits, four for β, two for γ, and one for α2 (Bech-Hansen et al., 1998; Letts et al., 1998; Ophoff et al., 1998; Strom et al., 1998). Expression of these α1 subunits led to the induction of typical HVA currents in terms of their biophysical and pharmacological properties. Along with these expected properties, some HVA channels exhibited properties that were once considered specific to T-type channels. Specifically, fast inactivation, inactivation at negative membrane potentials, and block by micromolar concentrations of nickel are no longer distinguishing features (Ellinor et al., 1993;Soong et al., 1993; Zamponi et al., 1996). However, T-type currents can still be distinguished from HVA currents by the following criteria: low voltage-activation, criss-crossing pattern of currents, slow deactivation, and tiny single-channel conductance (Matteson and Armstrong, 1986; Carbone and Lux, 1987; Fox et al., 1987; Randall and Tsien, 1997).
Recently our lab has published the cloning and expression of two new α1 subunits, α1G and α1H, that display all the characteristic features of T-type currents (Cribbs et al., 1998; Perez-Reyes et al., 1998). In this study we report the cloning of a third member of this T-type channel family, α1I. We compared its electrophysiological properties to those of the cloned T-type channels α1G and α1H and to the high voltage-activated channels formed by α1Eβ3. Based on its brain distribution and novel gating properties, we suggest that α1I plays important roles in determining the electroresponsiveness of neurons.
MATERIALS AND METHODS
cDNA library screening. A rat brain λgt10 cDNA library (catalog #RL3005a; Clontech, Palo Alto, CA) was screened using conventional filter hybridization according to the manufacturer’s protocol. All cDNA probes were released from the vector by restriction digestion, separated on agarose gels, and purified using the Qiaquick gel extraction kit (Qiagen, Valencia, CA). Probes were labeled using 32P-α-dCTP and the RadPrime DNA labeling system (Life Technologies, Grand Island, NY). Probes were derived from either Integrated Molecular Analysis of Genomes and their Expression (IMAGE) Consortium (LLNL) clones (Lennon et al., 1996) (ID numbers 402278 and 50902; obtained from Genome Systems, St. Louis. MO) or PCR products. PCR primers were designed from either partial clones (IIS1f, N45) or from the genomic clone 206C7 (GenBank accession number AL008716; direct submission by J. Burgess, Wellcome Trust Genome Campus, Cambridgeshire, UK). The PCR primer sequences were as follows: IS1f, TGC ACG TGG TTT GA(AG) TG(TC) GT; IIS2r, GGC CAG CTT CAG (GAT)AT CAT (CT)TC; IIS1f, ATG GCT ATC CTG GTG AAC AC; IIIS1r, TGG GCA ATG ATG GT(CT) TG(AG) CA; IIIS1f, TTC CGG GTC CTG TG(TC) CA(AG) AC; and N45, GAT GAT GGT GGG (AG)TT GAT. The primers are named according to their approximate location in the protein and their direction (f, forward; r, reverse). The full-length construct was assembled from five cDNA clones (Fig.1A) in the vector pGEM-HEA (a modified version of pGEM-HE; Liman et al., 1992; gift from Kenton Swartz, National Institutes of Health, Bethesda, MD). This vector contains 5′ and 3′ untranslated regions from aXenopus β globin gene. Because of poor growth of bacterial cultures (INVαF′; Invitrogen, Carlsbad, CA) transformed with this construct, we recloned the full-length cDNA into pSP73 (Promega, Madison, WI) along with the 5′ globin sequence [vector coordinates,KpnI (26)/XmaI (89)]. The same full-length cDNA was also subcloned into pcDNA3 (Invitrogen) for expression in mammalian cells. The sequence of α1I was determined on both strands of the plasmid using oligonucleotide primers, Sequenase 2.0 (Amersham, Arlington Heights, IL), a digitizer, and WDNASIS software (Hitachi, San Bruno, CA). Regions of compressed sequence were resolved using the 7-deaza-GTP sequencing reaction mix (Amersham).

Primary structure and predicted topology of the rat α1I (CavT.3). A, Schematic showing the location of the restriction enzyme sites and clones used for constructing the full-length cDNA. The cDNA construct was assembled from the following clones: λgt10 clone RF17, NgoM1 (−124)/AvrII (1354); PCR clone number 13,AvrII (1354)/BglII (1893); PCR clone number 2, BglII (1893)/BamHI (3357); a synthetic pair of oligonucleotides, BamHI (3357)/HindIII(3386); PCR clone b,HindIII(3386)/ApaLI (4327); and λgt10 clone ME4, ApaLI (4327)/EcoRI (polylinker). B, Deduced amino acid sequence of the rat α1I T-type calcium channel. Residues conserved among the rat α1I, rat α1G, and the human α1H are shown in capitalized bold letters. Putative membrane-spanning regions are markedabove the sequence. Analysis of the α1I protein sequence with a modified Prosite database identified the following: four cAMP-dependent protein kinase phosphorylation motifs (R/K-R/K-x-S/T or R/K-R/K-x-x-S/T) all located in the intracellular loops (marked above site with the lettera); 18 protein kinase C motifs (S/T-x-R/K), eight of which are located intracellularly (marked with c); one tyrosine phosphorylation motif (R/K-x-x-x-D-x-x-Y) located at the start of IIS1 (marked with y), and seven N-linked glycosylation motifs (N-x-S/T), five of which are in extracellular loops (marked with n). C, Schematic of the α1I channel showing relationship of loops to the plasma membrane. Each amino acid residue is represented by a circle. For diagrammatic purposes, the membrane-spanning regions are modeled as α helices.
Northern analysis. Northern blots of 2 μg of mRNA were obtained from either Origene (Rockville, MD) or Clontech. The blots were hybridized at 42°C for 16–20 hr in standard solutions (Sambrook et al., 1989) containing 50% formamide. Blots were washed up to 65°C in a final buffer of 0.1× SSC (15 mm NaCl and 1.5 mm Na citrate) and 0.1% SDS, then exposed to x-ray film (Hyperfilm MP; Amersham) at −80°C between two intensifying screens. The probe was an NcoI fragment (nucleotides 5142–6197) of clone ME4, which includes the last 363 bp of the coding region and 692 bp of 3′ untranslated region; none of this sequence is found in either α1G or α1H.
Oocyte expression. Capped cRNA was synthesized from plasmid linearized with EcoRI using T7 RNA polymerase (Ambion, Austin, TX). The concentration of cRNA was measured spectrophotometrically. Oocytes were prepared from Xenopus laevis (Xenopus One, Ann Arbor, MI) using standard techniques (Leonard and Snutch, 1991). Each oocyte was injected with 2–10 ng of cRNA in a volume of 50 nl. The results were obtained from five batches of oocytes derived from five frogs.
Electrophysiological analysis of injected oocytes. Oocytes were voltage-clamped using a two-microelectrode voltage-clamp amplifier (model OC-725B; Warner Instrument, Hampden, CT). The standard bath solution contained the following (in mm): 10 Ba(OH)2, 90 NaOH, 1 KOH, 0.1 EDTA, and 5 HEPES, adjusted to pH 7.4 with methanesulfonic acid. Voltage and current electrodes (1.5–1.8 MΩ tip resistance) were filled with 3m KCl. Except where noted, data were filtered at 1 kHz (model 902 filter; Frequency Devices, Haverhill, MA) and digitized at 4 kHz using the pClamp system (Digidata 1200 and pClamp 6.0; Axon Instruments, Foster City, CA). In some experiments, oocytes were injected with 50 nl of 25 mm BAPTA (Molecular Probes, Eugene, OR). Oocytes were allowed to recuperate for at least 1 hr but not more than four.
For single-channel recording, the vitelline membrane was removed with forceps after shrinking in hypertonic media (120 mmK+-aspartate, 25 mm KCl, 1 mm MgCl2, 10 m EGTA, and 10 mm HEPES, pH 7.4) (Methfessel et al., 1986). Oocytes were then transferred to a depolarizing bath solution containing (in mm): 120 K+-glutamate, 25 KCl, 1 Ca2+-ATP, 2 EGTA, 10 glucose, and 10 HEPES, pH 7.4 (Lacerda et al., 1994). Pipettes were made from 7052 glass tubing, and the tips were coated with Sylgard 184 (Dow Corning, Midland, MI). The pipette solution contained (in mm): 115 BaCl2, 1 EGTA, and 10 HEPES, pH 7.4 (Lacerda et al., 1994). Single-channel currents were acquired at 10 kHz and filtered at 2 kHz using an Axopatch 200B, a Digidata 1200 interface, and pClamp 5 software.
Generation of stably transfected human embryonic kidney-293 cells. Human embryonic kidney-293 (HEK-293) cells (1 × 106 cells in a 100 mm culture dish) were transfected with 10 μg of cDNA of either α1I, rat α1G (Perez-Reyes et al., 1998), human α1H (Cribbs et al., 1998), or human α1E (Schneider et al., 1994) plus human β3 (Murakami et al., 1996; a gift from V. Flockerzi). Forty-eight hours after transfection, the cells were suspended in DMEM medium supplemented with G418 (1 gm/l, Life Technologies), 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. Individual colonies were isolated with cloning rings. Results were obtained using the following cell lines: α1G, Nr2+; α1H, number 13; and α1Eβ3number 1C5. The results from three distinct α1I-transfected cell lines (numbers 11, 19, and 25) were identical and have been pooled.
Electrophysiological analysis of HEK-293 transfected cells.HEK-293 cells were dissociated by digestion with 0.25% trypsin plus 1 mm EDTA (Life Technologies) for 2 min, then diluted 20-fold with DMEM. The cells were triturated, diluted twofold with DMEM, then plated on coverslips. The cells were incubated at least 4 hr and up to 2 d before electrophysiological studies. The internal pipette solution contained the following (in mm): 55 CsCl, 75 CsSO4, 10 MgCl2, 0.1 EGTA, and 10 HEPES, pH adjusted to 7.2 with CsOH. The recording solution contained the following (in mm): 10 BaCl2 solution (or 2 CaCl2), 140 tetraethylammonium (TEA) chloride, 6 CsCl, and 10 HEPES, pH adjusted to 7.4 with TEA-OH. Whole-cell currents were recorded from ruptured patches using an Axopatch 200A amplifier, Digidata 1200 analog-to-digital converter, and pClamp 6.0 software (Axon Instruments). Data were filtered at 1 kHz and digitized at 2 kHz, except for measurement of tail currents that were digitized at 50 kHz. Pipettes were made of TW-150–6 capillary tubing, using a model P-97 Flaming–Brown pipette puller (Sutter Instruments, Novato, CA). Under these solution conditions, the pipette resistance was typically 1.5–2.0 MΩ. Series resistance (correction and prediction) and cell capacitance were compensated ∼80%. The average cell capacitance was ∼25 pF. The data were not corrected for any residual leak currents. All experiments were performed at room temperature.
Data analysis. Peak currents, integrals, and exponential fits to the electrophysiological data were determined using Clampfit software (Axon Instruments). Conductance was calculated using the Goldman–Hodgkin–Katz equation (Hille, 1992) and the solver function of Excel (Microsoft). Capacitative transients were removed from single-channel records by subtracting null sweeps taken from the same experiment and were then analyzed using Transit (VanDongen, 1996). Single-channel amplitudes were measured by averaging the values obtained from both Gaussian fits to all-points histograms of traces with openings and amplitude histograms of all idealized openings. Fits and graphing of the data were with Prism (GraphPad, San Diego, CA).
RESULTS
Cloning of T-type Ca2+ channels began with the identification of an EST clone [IMAGE Consortium (Lennon et al., 1996) clone ID number 44039] as being derived from a novel channel (Perez-Reyes et al., 1998). Homology analysis of the full sequence of this clone (GenBank accession number AF02922) identified aCaenorhabditis elegans homolog (GenBank accession number 1017809). The amino acid sequence corresponding to the sixth membrane-spanning region of repeat IV (IVS6) was used to search the EST database using the BLAST algorithm (Altschul et al., 1990), leading to the identification of IMAGE Consortium clones number 50902 (GenBank accession number H19230) and number 402278 (GenBank accession numberW76774). Subsequent cloning of the full-length cDNAs for α1G and α1H and mapping of their chromosomal location allowed us to identify these clones as being derived from two genes, CACNA1G andCACNA1H (Cribbs et al., 1998; Perez-Reyes et al., 1998).
A rat brain cDNA library was screened at low stringency with H19230(α1G), leading to the isolation of fifty positive plaques. Many of these plaques were not detected in the secondary screening. To test if these lost plaques were derived from α1H, they were rescreened withW76774. Two recombinants were detected with this probe and plaque-purified (clones ME4 and ME5). Sequencing of their cDNA inserts demonstrated that they were similar to each other but clearly different from either α1G or α1H, hence, we called it α1I or CavT.3. This conclusion was supported by having a representative member of each gene cloned from the rat brain library (UN7, α1G; ME3, α1H; and ME4, α1I). A subsequent search of the HTGS division of the GenBank with the full-length α1G sequence (AF027984) allowed us to recognize the human genomic sequence of α1I (and refer to the gene as CACNA1I) on cosmid 20CC7 derived from chromosome 22. This sequence was used to design three sets of PCR primers to clone the cDNA encoding repeats I-III.
The PCR product containing repeat I was used to isolate the 5′ end from the rat brain λgt10 cDNA library. Four clones were isolated, but only RF17 extended into the presumptive 5′ untranslated region. Although clone RF17 contains 278 base pairs at the 5′ end that are enriched with the nucleotides G and C (80% compared with 58% for the coding region), it does not contain an in-frame stop codon. Additional support for our assignment of the start codon comes from the sequence of human cosmid clone 1104E15 (direct submission by J. Sulton, Wellcome Trust Genome Campus). This clone contains a presumptive exon encoding 79 amino acids that are 83% identical to the rat sequence, spanning from the amino terminus sequence to IS1. An in-frame stop codon occurs 171 bp before the start codon we predicted from the rat sequence.
The full-length rat α1I cDNA is composed of 6503 bp (GenBank accession number AF086827). The open reading frame covers 5505 bp, encoding a protein with a predicted molecular weight of 205,198 (Fig.1B). The α1I protein is 59.3% identical to human α1H and 56.9% identical to rat α1G. In contrast, it is only 13–19% identical to the HVA α1 subunits. Most of the residues conserved in all three T channel proteins (Fig. 1B) and in HVA α1 subunits are found in the putative membrane-spanning regions. These membrane-spanning regions also share considerable structural homology to voltage-gated K+ and Na+ channels (Jan and Jan, 1990), suggesting that the overall topology (Fig. 1C) of these channels is similar (Durell et al., 1998). The intracellular loops connecting each repeat and the C terminus are poorly conserved. The T channel α1 proteins contain stretches of histidine and arginine residues, as noted previously for high voltage-activated α1 subunits (Perez-Reyes and Schneider, 1994). In α1G and α1H, this motif occurs in the I-II linker, whereas in α1I it occurs in the II-III linker. In contrast to HVA α1 subunits, the three LVA channels contain a large (107 residues) extracellular loop located between IS5 and the P loop. All other extracellular loops are predicted to be smaller (<35). Although the amino acid sequence is not highly conserved (45%), there are six conserved cysteine residues. Because disulfide bonds are formed in extracellular domains of proteins, this loop may play a role in localizing channels to the cell surface. The C terminus is composed of only 143 amino acids, which is similar in length to human α1H (185), but shorter than either rat α1G (430) or HVA channels such as human α1C (773) or α1E (590). It also contains nine sequential copies of the repeat, TGCCCC, leading to runs of prolines and cysteines. This repetitive element is not found in the human genomic sequence. Analysis of the α1I protein sequence with a modified Prosite database identified the following: four cAMP-dependent protein kinase phosphorylation motifs located in the intracellular loops (Fig.1B), eight protein kinase C motifs located intracellularly, one tyrosine phosphorylation motif located at the start of IIS1, and five N-linked glycosylation motifs on extracellular loops. Motifs for binding β subunits of either G-proteins (Q-x-x-E-R;Chen et al., 1995) or HVA calcium channels (Q-Q-x-E-x-x-L-x-G-Y-x-x-W-I-x-x-x-E; DeWaard et al., 1996) were not identified.
The distribution of α1I mRNA in various rat tissues was determined by Northern blot analysis (Fig. 2). The predominant species detected had a mobility corresponding to 10.5 kb and was only found in brain. Similar results were obtained with two other blots. Minor bands were also observed at 2 and 8 kb. The intensity of the 2 kb band varied between experiments and showed a wider tissue distribution.

Distribution of α1I mRNA by Northern blot analysis. A rat multiple-tissue blot was probed with32P-labeled α1I (nucleotides 5142–6197) and exposed for 5 d. Size markers are indicated on the right in kilobases. The faint 8 kb bands in kidney and liver were only observed in one experiment and may be caused by contamination of the probe with sequence encoding repeat IV leading to cross-hybridization with α1H (Cribbs et al., 1998). Alternatively, these bands may represent cross-hybridization with a distinct mRNA.
Functional expression of α1I currents was first studied inXenopus oocytes injected with cRNA. Quite surprisingly, the currents activated very slowly, particularly at threshold voltages where the time-to-peak was >150 msec (Fig.3A,B). Robust expression (>1 μA) was obtained in most batches of oocytes. Oocytes expressing >2 μA were excluded from the analysis, which reduced the average peak current to −718 ± 146 nA. Kinetics were not affected by BAPTA injection into the oocytes before recording (n = 5), so the data were pooled. This result indicated that there was minimal activation of the Ca2+-activated Cl current. To investigate the possibility of a mutation, the full-length cDNA construct was sequenced. No striking differences were observed in the sequence of α1I as compared with either the human genomic sequences containingCACNA1I, (GenBank accession numbers AL022319, AL008716), rat α1G (GenBank accession number AF027984), or human α1H (GenBank accession number AF051946).

Comparison of the α1I currents to cloned α1G, α1H, and α1Eβ3 channel currents. Currents were evoked by step depolarizations to varying test potentials from a holding potential of −90 mV. Currents were measured in stably transfected HEK-293 cells using the ruptured patch-clamp method with 10 mm Ba2+ as the charge carrier. Also shown are results from Xenopus oocytes expressing α1I.A, α1I currents expressed in HEK-293 cells andXenopus oocytes were compared with α1G and α1H currents expressed in HEK-293 cells. Currents from the peak of the current–voltage relationship have been scaled and superimposed. Data were taken from the same cells shown in panelsB–F. B, Representative current traces recorded from oocytes injected with α1I-cRNA. Currents were evoked during test pulses that incremented 7 mV with each episode.C–F, Representative currents from HEK-293 cells stably transfected with either α1I (C), α1G (D), α1H (E), or α1Eβ3(F). Currents were elicited by depolarizing 10 mV steps from −90 mV.
When α1I was expressed by stable transfection into HEK-293 cells, the currents were twofold faster (80 msec time-to-peak at threshold; Fig.3A,C) than observed in oocytes, but their kinetics were much slower than observed previously for either α1G (Perez-Reyes et al., 1998) or α1H (Cribbs et al., 1998). To compare the gating properties of cloned voltage-gated Ca2+ channels, we prepared stably transfected HEK-293 cells of α1I, α1G, α1H, and α1E plus β3. Robust expression (>1 nA; Fig.4A) was obtained with all cloned channels. Representative current traces obtained during pulses of varying test potentials are shown in Figure3C–F. The peak currents were averaged and plotted versus test potential (Fig. 4A). To illustrate the position of these current–voltage curves, the data from each cell were normalized to the largest peak current observed, then averaged (Fig. 4B). These results indicated that α1I, α1G, and α1H channels were all activated at low voltages. In contrast, α1Eβ3 channels required stronger depolarizations (30 mV) to open. To quantitate these differences, conductance was calculated using the Goldman–Hodgkin–Katz equation and fit with the Boltzmann equation (see Fig. 6C). The values of half-maximal activation (V0.5) and slope (k) are presented in Table 1. These results show that each cloned T-type channel activated at slightly different potentials, with α1H being the most negative followed closely by α1G, whereas α1I activated at 7 mV higher test potentials. In contrast, α1Eβ3 currents activated 15 mV more positive than α1I. These results were obtained using 10 mm Ba2+ as the charge carrier. Because of the effects on surface charge screening by such high concentrations of divalent cation (Wilson et al., 1983), we also measured currents under more physiological conditions (2 mmCa2+; Table 1). The currents through α1I, α1G, and α1H channels also had an apparent reversal potential that was ∼15 mV more negative than α1Eβ3 (Fig.4B). Integrated currents from representative cells, each of which had ∼1 nA of peak current, were also plotted as a function of the test potential (Fig. 4C). These results showed that α1I caused the biggest influx of Ba2+among the cloned channels.

Comparison of the current–voltage (I–V) relationships of α1I to those of α1G, α1H, and α1Eβ3. Symbols representing each cloned channel are the same in Figures 4-6: α1G (▵), α1H (▿), α1I (○), and α1Eβ3 (▪). A, Average peak currents elicited during test pulses to the indicated potentials. Data represent the mean ± SEM from the following number of cells: α1G (n = 8), α1H (n = 6), α1I (n = 10), and α1Eβ3(n = 10). B, The data inA were normalized to the peak current observed for each cell then averaged. Also shown is the average data obtained with oocytes injected with α1I (●; n = 12).C, Integral of the current measured during each test pulse is plotted as a function of test potential. Representative cells were chosen that each expressed 1 nA current at the peak of theI–V.
Summary of the voltage-dependent properties of cloned calcium channels
To measure both activation and inactivation time courses, the pulse was lengthened to 350 msec, and the resulting data were fit with two exponentials (Fig.5A,B). As observed for both native (Huguenard, 1996) and cloned T-type channels (Cribbs et al., 1998; Perez-Reyes et al., 1998), activation and inactivation kinetics were slow near threshold voltages and accelerated with increasing depolarizations, producing a classical criss-crossing pattern (Randall and Tsien, 1997). This pattern was clearly distinct from HVA channels such as α1Eβ3 (Fig.3F) whose activation and inactivation time constants were relatively voltage-independent (Fig.5A,B). The current kinetics of α1G and α1H were voltage-dependent and were nearly identical to each other. Similar results were obtained previously with α1G expressed in oocytes (Perez-Reyes et al., 1998) and α1H in transiently transfected HEK-293 cells (Cribbs et al., 1998). In contrast, α1I kinetics were threefold slower in HEK-293 cells and sixfold slower in oocytes.
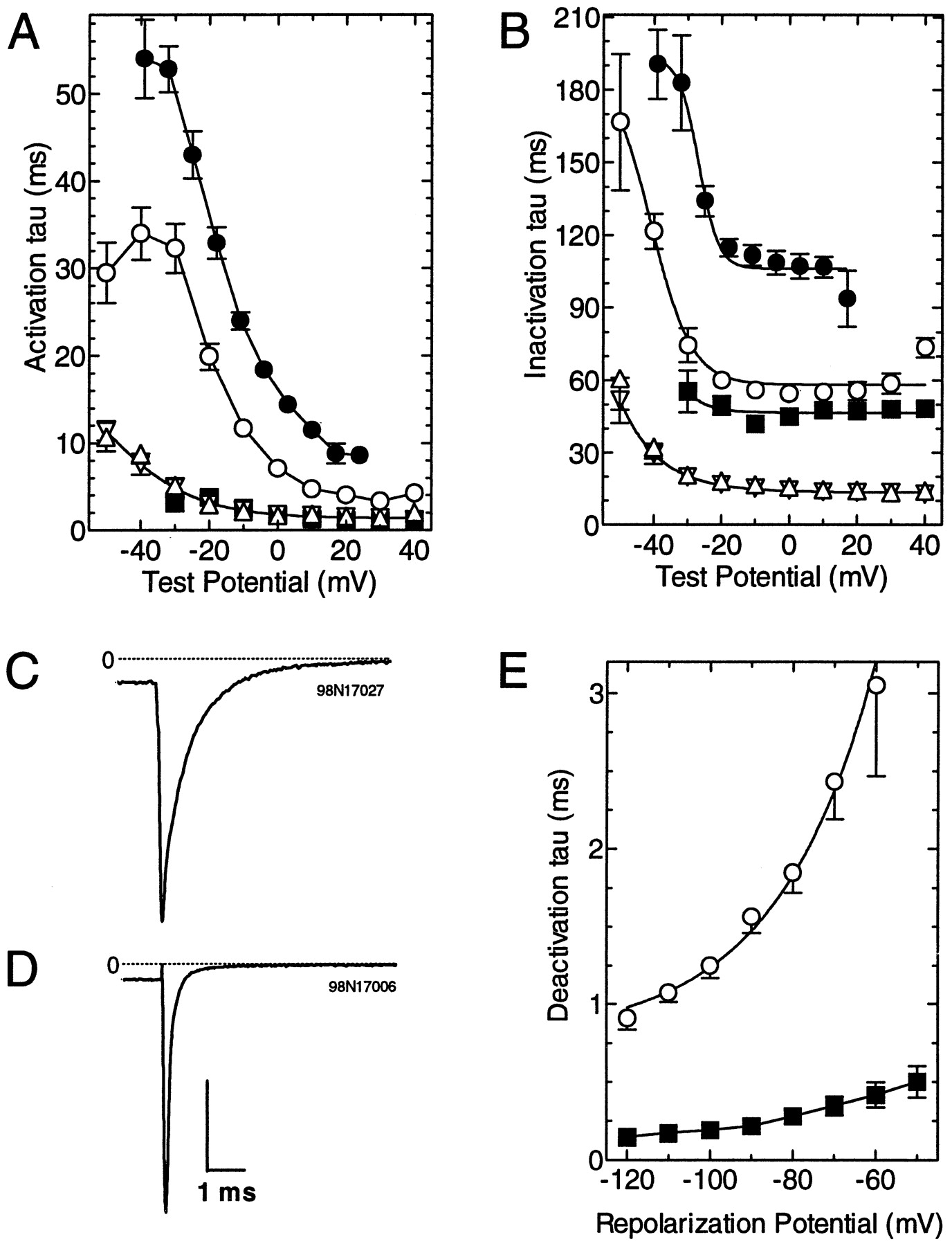
Comparison of the kinetic properties of α1I with those of α1G, α1H, α1I, and α1Eβ3.A, B, Currents elicited during theI–V protocol were fit with two exponentials. Average activation (A) and inactivation (B) tau values are plotted as a function of test potential. All currents were recorded from HEK-293 cells, except for the data represented by ●, which are from oocytes injected with α1I. Data represent the mean ± SEM from the following number of cells: α1G (▵, n = 8), α1H (▿,n = 6), α1I (○, n = 10), α1I in oocytes (●, n = 15), and α1Eβ3 (▪, n = 14).C, D, Representative tail currents from cells expressing either α1I (C) or α1Eβ3 (D). Currents were evoked by test pulses to either −20 (α1I) or 0 (α1Eβ3) mV, followed by repolarization to −100 mV. Vertical scale bar represents 1 (C) or 5 nA (D).E, Data obtained in C andD were fit with a single exponential. Average deactivation time constants of α1I (n = 4) and α1Eβ3 (n = 4) tail currents were plotted as a function of repolarization potential.
A second defining feature of T-type Ca2+ currents is that they deactivate relatively slowly, producing slowly decaying tail currents after a depolarizing pulse. Representative tail currents for α1I and α1Eβ3 are shown in Figure 5, C andD, respectively. The data were fit with a single exponential to determine the time constant for deactivation (Fig. 5E). These results indicated that α1I channels closed at least sixfold slower than α1Eβ3 channels (at −100 mV, α1I τ, 1.25 ± 0.08 msec; α1Eβ3, 0.19 ± 0.03 msec; n = 4 for both). Fast deactivating tail currents have also been reported previously for both α1Eα2β1a and R-type currents (Williams et al., 1994; Randall and Tsien, 1997).
Inactivation was also studied by applying 5-sec-long prepulses that were terminated by a brief (5 msec) repolarization to close any open channels, then followed by a test pulse to −30 mV to measure channel availability. Representative current traces recorded during prepulses to −50 and −55 mV are shown in Figure6A. Average data were fit with the Boltzmann equation (Fig. 6B, Table 1). These results showed that each cloned T-type channel inactivated at slightly different potentials, with α1H inactivating at the lowest potentials, followed by α1G, whereas α1I required potentials that were 15 mV higher. Comparison of α1I channels in HEK-293 cells to those expressed in oocytes indicated that the voltage dependence of activation was nearly identical (Fig. 4B) but that inactivation occurred at 7 mV higher potentials in oocytes (V50 = −57.7 ± 0.8; n = 7). In contrast, the voltage dependence of α1G was nearly identical in HEK-293 cells, as reported previously for oocytes (Perez-Reyes et al., 1998). The traces in Figure 6A were chosen to illustrate that there are voltages at which channels were activated during the prepulse, but they were not completely inactivated, as evidenced by currents evoked during the test pulse. This activity is referred to as a window current and is typically illustrated by the overlap in the steady-state inactivation and activation curves. The resulting window regions for α1I, α1G, α1H, and α1Eβ3 are shown in panels D–I of Figure 6. Of the three T-type channels, α1I had the largest window region. In contrast, α1Eβ3 currents did not display a significant window region because inactivation occurred at very negative potentials. The window region is also shown for α1I currents measured with 2 mm Ca2+ (Fig.6I). At the peak of the window (−64 mV), ∼0.6% of the α1I channels may open (percentage of channels available to gate times the number of channels that gate at that potential).

Comparison of steady-state inactivation, activation, and window currents of α1I to those of α1G, α1H, and α1Eβ3. A, The voltage protocol used to measure inactivation is shown above representative traces obtained during prepulses to −50 and −55 mV. The protocol also included a short 5 msec repolarization to −90 mV at the end of the prepulse. The time between episodes was 15 sec. B, Average percent inactivation was plotted as a function of prepulse voltage. The average data were fit with the Boltzmann equation (smooth curves). Data represent the mean ± SEM from the following number of observations: α1G (▵,n = 6), α1H (▿, n = 8), α1I (○, n = 7), and α1Eβ3 (▪,n = 12). C, Conductance was calculated using the Goldman–Hodgkin–Katz equation. The data were averaged, then fit with the Boltzmann equation (smooth curves). Data represent the mean ± SEM from the following number of observations: α1G (n = 8), α1H (n = 6), α1I (n = 8), and α1Eβ3 (n = 14).D–I, Activation and inactivation curves shown inB and C were overlapped and expanded to show window currents. Data for α1I (D), α1G (E), α1H (F), α1Eβ3 (G), and α1I expressed in oocytes (H) were recorded in 10 mm Ba2+ solutions. Also shown are data obtained using 2 mm Ca2+ as the charge carrier from HEK-293 cells stably transfected with α1I (I). Smooth curvesrepresent Boltzmann fits to the all the activation data points and to inactivation data points that were >50%.
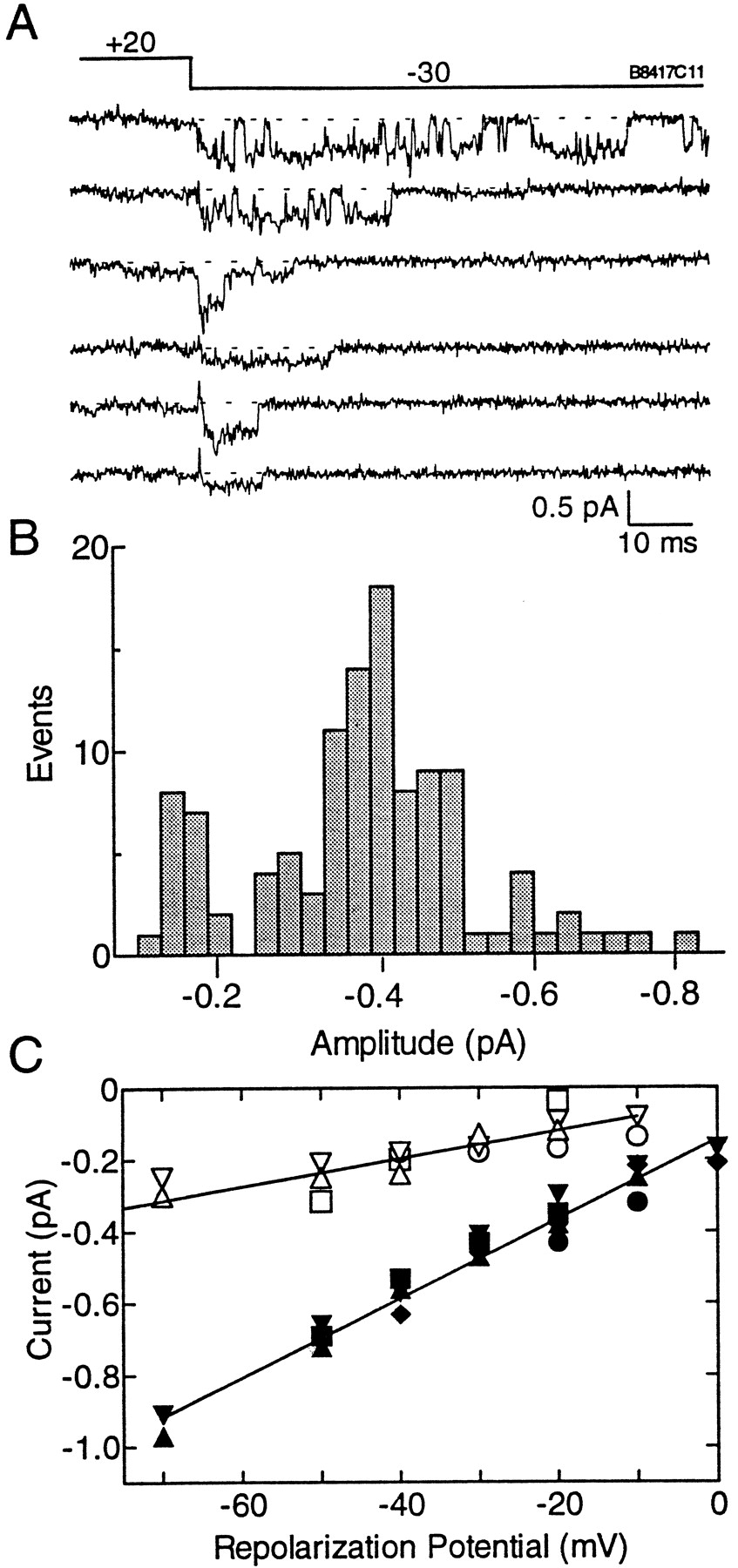
T-type channels are also defined by their tiny single-channel conductance in saturating concentrations of Ba2+(Fox et al., 1987; Huguenard, 1996). To measure these small currents, we used a tail current protocol that increases the probability of channel opening at negative potentials where the driving force is larger, and hence the currents are larger. Representative sweeps were chosen to illustrate that channel openings occur in bursts and to show the presence of a subconductance state (Fig.7A). The third trace in Figure7A shows a channel closing from the full to a subconductance state, whereas traces 4 and 6 show openings to the subconductance state. The data were idealized with the Transit algorithm (VanDongen, 1996), and the amplitude of the idealized openings were plotted in Figure 7B. This plot showed that channels opened to two distinct amplitudes. Gaussian fits to these histograms were used to determine the amplitude of the openings, then plotted as a function of repolarization potential. The data were then fit by linear regression to determine the slope conductance. The conductance of the small openings was 3.9 ± 0.5 pS, whereas the larger openings had a conductance of 11.0 ± 0.5 pS. The current at 0 mV for the full conductance state was −0.15 pA.

Single-channel currents of α1I measured fromXenopus oocytes using the cell-attached patch-clamp method. A, Representative traces from a single patch displaying full and subconductance openings of α1I. The voltage protocol contained a prepulse to +20 mV followed by a test pulse to −30 mV. B, Channel openings and closings were idealized using Transit to determine the amplitude of channel openings. Data are taken from the same patch shown in A. C, Single-channel conductance of α1I currents. The amplitudes of single channels were obtained from Gaussian fits to amplitude histograms of idealized openings. The amplitudes were plotted against test potential. Slope conductances were calculated by linear regression through all the data points. Data were obtained from five patches on four oocytes.Filled symbols represent full conductance states, whereas open symbols represent subconductance states measured from the same patch.
DISCUSSION
The present study describes the cloning and expression of a novel member of the T-type Ca2+ channel family. Discovery of this family of genes was made possible by the development of normalized cDNA libraries (Soares et al., 1994), systematic sequencing of clones from these libraries, and free access to their DNA sequences (Lennon et al., 1996). We cloned the first cDNA fragment of α1I by low-stringency screening of a rat brain cDNA library with two IMAGE Consortium clones that encoded either α1G or α1H. The cloning project was greatly facilitated by efforts to sequence the human genome, in particular the work performed by the Wellcome Trust Genome Campus, which led to partial sequencing of the human α1I gene,CACNA1I, and its localization on human chromosome 22q12.3–13.2.
Sequence homology can be used to subdivide the family of voltage-gated Ca2+ channel α1 subunits into three subfamilies: (1) T-type (G, H, I); (2) L-type (S, C, D, F); and (3) non-L-type HVA (A, B, E). Electrophysiological characterization of these cloned channels has revealed considerable functional differences between the members of each subfamily. For example, α1S encodes a slow Ca2+ channel (Perez-Reyes et al., 1989), whose main physiological role is as a voltage sensor, coupling depolarization to skeletal muscle contraction (Stern et al., 1997). Similarly α1E has unique characteristics of inactivation gating and permeation that set it apart from α1A and α1B (Soong et al., 1993; Bourinet et al., 1996). In the T-type subfamily, it is α1I that stands apart. It encodes a slowly activating and inactivating Ca2+channel that gates in voltage ranges similar to, but higher than the other two cloned T channels. It can be classified as a T-type channel by virtue of its slowly deactivating tail currents and tiny single-channel conductance in 115 mm BaCl2.
The deduced amino acid sequence of α1I is ∼58% identical to either α1G or α1H, but only ∼15% identical to the HVA α1 subunits. The regions of least conservation between the T-type channels are their intracellular loops. The III-IV linker is an exception because it is 75% identical among the three T channel proteins. Perhaps this high degree of sequence identity is caused by conservation of function as observed in voltage-gated Na+ channels where the III-IV linker plays a role in fast inactivation (Catterall, 1995). A role of the intracellular linkers in inactivation was postulated before the structure of T channels was even deduced (Miller and Hu, 1995).
Three major conclusions from our expression studies with α1I are: one, it encodes a T-type Ca2+ channel with a unique voltage dependence; two, it encodes a slow channel; and three, its activity is dependent on the expression system. Expression in both heterologous expression systems demonstrated that α1I encoded low voltage-activated currents. The threshold voltage for channel activation was −60 mV (in 10 mm Ba2+), which was similar to what we have observed for α1G and α1H (Cribbs et al., 1998; Perez-Reyes et al., 1998). Expression inXenopus oocytes led to α1I currents that were as slow as those observed for the L-type channels of skeletal muscle (Garcia et al., 1992). In contrast, α1I currents from transfected HEK-293 cells activated and inactivated much more quickly. In addition, the voltage dependence of steady-state inactivation differed between these two expression systems, with the oocyte currents requiring 8 mV higher depolarizations. The reason for this discrepancy is under investigation. A plausible explanation is that HEK-293 cells, or oocytes, express a subunit of T-type channels that can influence kinetics and steady-state inactivation. High voltage-activated Ca2+ channels are multisubunit complexes, which in addition to α1 subunits, also contain at least two and sometimes three auxiliary subunits, α2δ, β, and γ. All of these subunits have been reported to modulate channel properties (Perez-Reyes and Schneider, 1994); by analogy, it is likely that LVA channels also have accessory subunits. It should be noted that HEK-293 cells were originally derived from human kidney (Graham et al., 1977), which is the tissue with the highest expression of α1H (Cribbs et al., 1998). It is also interesting to speculate that the newly identified γ2 subunit may be a T-type channel subunit (Letts et al., 1998). Mutations in the γ2 gene are thought to be responsible for the absence epilepsy phenotype of the stargazer mouse. Similarly, it has been suggested that increased T channel activity may cause absence epilepsy in rats (Tsakiridou et al., 1995).
Three criteria can be used to define T-type channels, their opening at membrane potentials near the resting membrane potential of most cells (LVA), their slow closing after a depolarization (SD), and their tiny (T) single-channel conductance in saturating concentrations of Ba2+ (Matteson and Armstrong, 1986; Carbone and Lux, 1987; Fox et al., 1987). T-type channels also have a distinctive criss-crossing set of current traces obtained during theI–V protocol (Randall and Tsien, 1997). Despite its slow kinetics, α1I still produces this distinctive pattern. This pattern is the result of the voltage-dependence of T channel kinetics, where activation kinetics are determined by the latency to first opening and by an inactivation process that is tightly coupled to activation (Carbone and Lux, 1987; Droogmans and Nilius, 1989; Chen and Hess, 1990; Miller and Hu, 1995). Despite the slower activation kinetics than either α1G or α1H, α1I deactivates with a similar time course, producing a slow tail current. HVA channels, such as α1E, close at least sixfold faster. The exact mechanism by which T channels, which have a mean open time of ∼1 msec, produce such a slow tail has not been fully characterized.
One of the early methods for separating LVA from HVA currents was to record currents from holding potentials of −90 and −40 mV, then subtract the currents. This was useful because in many cells only LVA currents inactivate at −40 mV. Recent studies suggest that some HVA channels inactivate at lower potentials than LVA channels, notably the R-type (Randall and Tsien, 1997) and the cloned α1E (Fig. 6). Therefore steady-state inactivation is no longer a defining feature of LVA channels. Among the cloned T channels, we find a 15 mV difference between the subtypes, with α1I requiring the highest prepulse potentials. Because activation of α1I is also shifted to more depolarized potentials, this leads to its having the largest window currents among the three cloned T channels. Notably, this window current occurs very close to the resting membrane potential of most cells, suggesting that α1I may play a role in determining resting concentrations of intracellular Ca2+. Window currents are an essential property of channels involved in pacemaker activity and play a critical role in the integration of synaptic potentials (Williams et al., 1997).
The single-channel conductance of native T channels ranges between 5 and 9 pS (Huguenard, 1996). Similarly, we found that α1G had a single-channel conductance of 7.5 pS and that α1H was slightly smaller, 5.3 pS (Cribbs et al., 1998; Perez-Reyes et al., 1998). The conductance of α1I was significantly larger (11 pS), approaching the value determined for rat α1E, 12.5 pS (Bourinet et al., 1996). In addition, rat α1E conducts Ba2+, Ca2+, and Sr2+ equally, as observed for native T channels (Shuba et al., 1991). Although α1E and α1I have similar slope conductances, the single-channel amplitudes are very different at 0 mV (α1E, −0.5 pA; α1I, −0.15 pA) because they have distinct reversal potentials. Evidence for the different reversal potentials was presented at the whole-cell level (Fig.4B). Measurement of the conductance of cloned T channels is complicated by the presence of a subconductance state. Evidence that these smaller openings are caused by the cloned T channels and not an endogenous oocyte channel was the following: (1) endogenous Ca2+ channels were not detected at the whole-cell level in these batches of oocytes, (2) endogenous channels generate −0.5 pA current at 0 mV (Lacerda et al., 1994), (3) transitions between the full and subconductance states are clearly visible (Fig. 7), and (4) both types of openings disappear when the holding potential is shifted to −40 mV (results not shown). The presence of subconductance states for native cardiac T channels (Droogmans and Nilius, 1989) and cloned HVA channels (Meir and Dolphin, 1998) have been noted.
The discovery of a clone encoding slow T-type channels may have been predicted from studies in native cells. Slow T-type channels have been described in neurons isolated from various rat thalamic nuclei, such as the reticular (Huguenard and Prince, 1992), laterodorsal (Tarasenko et al., 1997), and lateral habenula (Huguenard et al., 1993). They have also been described in a dorsal root ganglion–neuroblastoma hybrid cell line (Dolphin, 1998). These native T currents inactivated with nearly identical time constants as observed for α1I (55 msec). We suggest that these slow T channels are encoded by α1I. Support for this hypothesis is provided by the expression of α1I mRNA in these same brain regions (Talley et al., 1999). Notably, α1I is abundantly expressed in the thalamic reticular nucleus and lateral habenula. In addition, slow thalamic T channels required stronger depolarizations for channel opening than the fast T currents (Huguenard and Prince, 1992; Tarasenko et al., 1997). Similarly, we find that α1I gates at less negative potentials than either α1G or α1H. One notable difference is that the slow thalamic T current inactivated at more negative potentials than the fast, whereas we find the opposite result. However, we also found that the voltage dependence of inactivation varied between expression systems, suggesting this property may be affected by auxiliary subunits. Injection of thalamic–hypothalamic mRNA into Xenopus oocytes has been reported to produce LVA channels (Dzhura et al., 1996). The relationship of these currents to α1I is not clear, because these currents inactivated much more slowly.
Knowledge of the distribution and functional properties of the three T channels should lead to a greater understanding of their physiological roles. T channels are thought to play a pacemaker role in the genesis of rebound burst firing which, through reciprocal connections, can lead to oscillations and resonance of neuronal circuits (Huguenard, 1996). Burst firing of thalamic T channels is thought to be important in the transition to sleep and in the pathophysiology of epilepsy (McCormick and Bal, 1997). The ability of many antiepileptics to inhibit T channel activity led to the hypothesis that they may be involved in epilepsy (Coulter et al., 1990). Support for this hypothesis came from the observation that T channel activity of thalamic reticular neurons was increased 50% in GAERS (Tsakiridou et al., 1995), a well defined rat model of absence epilepsy (Vergnes and Marescaux, 1994). Cloning of T channels and the ability to express these channels at high density in stably transfected cells should provide an assay to study the pharmacological properties of T channels and may lead to the development of a new generation of antiepileptic drugs.
Footnotes
This work was supported by a grant from the National Institutes of Health to E.P.-R. (HL57828). We thank Qun Jiang for technical assistance.
Correspondence should be addressed to Edward Perez-Reyes, Department of Physiology, Loyola University Medical Center, 2160 South First Avenue, Maywood, IL 60153.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}