Sanfilippo Syndrome: Molecular Basis, Disease Models and Therapeutic Approaches



Abstract
:1. Introduction
2. Sanfilippo Syndrome
2.1. Subtype A
2.2. Subtype B
2.3. Subtype C
2.4. Subtype D
3. Heparan Sulfate
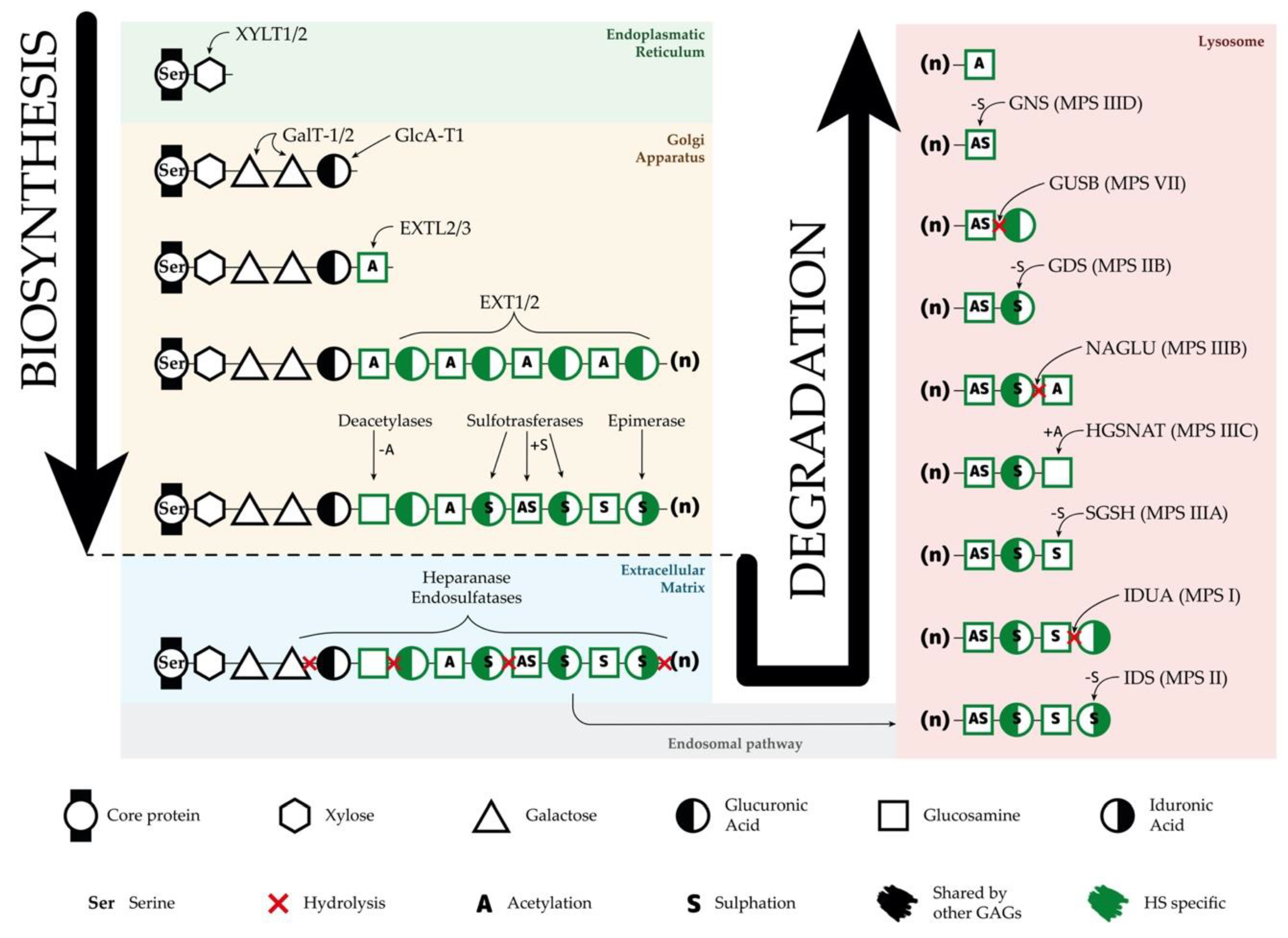
3.1. Heparan Sulfate Biosynthesis
3.2. Heparan Sulfate Degradation
3.3. Heparan Sulfate Accumulation and Disease Mechanisms
4. Disease Models
4.1. Animal Models
4.2. Cellular Models
5. Therapeutic Approaches
5.1. Enzyme Replacement Therapy
5.2. Substrate Reduction Therapy
5.3. Pharmacological Chaperones for Enzyme-Enhancement-Therapy
5.4. Stem Cell Therapy
5.5. Gene Therapy
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAV | Adeno-associated virus |
| BMP4 | Bone morphogenic protein 4 |
| CNS | Central nervous system |
| ER | Endoplasmic reticulum |
| ERT | Enzyme replacement therapy |
| FGF | Fibroblast growth factor |
| GAG | Glycosaminoglycan |
| GlcA | Glucuronic acid |
| GlcNAc | N-acetylglucosamine |
| GPC | Glial restricted progenitor cell |
| HS | Heparan sulfate |
| HSPG | Heparan sulfate proteoglycan |
| IdoA | Iduronic acid |
| iPSC | Induced pluripotent stem cell |
| LSD | Lysosomal storage disorder |
| MPS | Mucopolysaccharidosis |
| NSC | Neural stem cell |
| RNAi | Ribonucleic acid interference |
| shRNA | Short hairpin ribonucleic acid |
| siRNA | Small interference ribonucleic acid |
| SRT | Substrate reduction therapy |
References
- Neufeld, E.F.; Muenzer, J. The Mucopolysaccharidoses. In The Metabolic and Molecular Bases of Inherited Disease; Scriver, C.R., Valle, D.L., Antonarakis, S., Ballabio, A., Beaudet, A.L., Mitchell, G.A., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 3421–3452. [Google Scholar] [CrossRef]
- Sanfilippo, S.J.; Good, R.A.; Podosin, R.; Langer, L. Mental Retardation Associated with Acid Mucopolysacchariduria (Heparitin Sulfate Type). J. Pediatr-Us 1963, 63, 837–838. [Google Scholar] [CrossRef]
- Andrade, F.; Aldamiz-Echevarria, L.; Llarena, M.; Couce, M.L. Sanfilippo syndrome: Overall review. Pediatr. Int. 2015, 57, 331–338. [Google Scholar] [CrossRef] [PubMed]
- De Pasquale, V.; Pavone, L.M. Heparan sulfate proteoglycans: The sweet side of development turns sour in mucopolysaccharidoses. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 165539. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Evans, E.; Haslett, L.J. The lysosomal storage disease continuum with ageing-related neurodegenerative disease. Ageing Res. Rev. 2016, 32, 104–121. [Google Scholar] [CrossRef] [PubMed]
- Winder-Rhodes, S.E.; Garcia-Reitbock, P.; Ban, M.; Evans, J.R.; Jacques, T.S.; Kemppinen, A.; Foltynie, T.; Williams-Gray, C.H.; Chinnery, P.F.; Hudson, G.; et al. Genetic and pathological links between Parkinson’s disease and the lysosomal disorder Sanfilippo syndrome. Mov. Disord. 2012, 27, 312–315. [Google Scholar] [CrossRef] [PubMed]
- Fedele, A.O. Sanfilippo syndrome: Causes, consequences, and treatments. Appl. Clin. Genet. 2015, 8, 269–281. [Google Scholar] [CrossRef] [Green Version]
- Kowalewski, B.; Heimann, P.; Ortkras, T.; Lullmann-Rauch, R.; Sawada, T.; Walkley, S.U.; Dierks, T.; Damme, M. Ataxia is the major neuropathological finding in arylsulfatase G-deficient mice: Similarities and dissimilarities to Sanfilippo disease (mucopolysaccharidosis type III). Hum. Mol. Genet. 2015, 24, 1856–1868. [Google Scholar] [CrossRef] [Green Version]
- Khateb, S.; Kowalewski, B.; Bedoni, N.; Damme, M.; Pollack, N.; Saada, A.; Obolensky, A.; Ben-Yosef, T.; Gross, M.; Dierks, T.; et al. A homozygous founder missense variant in arylsulfatase G abolishes its enzymatic activity causing atypical Usher syndrome in humans. Genet. Med. 2018, 20, 1004–1012. [Google Scholar] [CrossRef] [Green Version]
- Coppa, G.V.; Galeotti, F.; Zampini, L.; Galeazzi, T.; Padella, L.; Santoro, L.; Maccari, F.; Gabrielli, O.; Volpi, N. Mild mental retardation and low levels of urinary heparan sulfate in a patient with the attenuated phenotype of mucopolysaccharidosis type IIIA. Clin. Biochem. 2013, 46, 688–690. [Google Scholar] [CrossRef]
- Meyer, A.; Kossow, K.; Gal, A.; Steglich, C.; Muhlhausen, C.; Ullrich, K.; Braulke, T.; Muschol, N. The mutation p.Ser298Pro in the sulphamidase gene (SGSH) is associated with a slowly progressive clinical phenotype in mucopolysaccharidosis type IIIA (Sanfilippo A syndrome). Hum. Mutat. 2008, 29, 770. [Google Scholar] [CrossRef]
- Moog, U.; van Mierlo, I.; van Schrojenstein Lantman-de Valk, H.M.; Spaapen, L.; Maaskant, M.A.; Curfs, L.M. Is Sanfilippo type B in your mind when you see adults with mental retardation and behavioral problems? Am. J. Med. Genet. C Semin. Med. Genet. 2007, 145C, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Ruijter, G.J.; Valstar, M.J.; van de Kamp, J.M.; van der Helm, R.M.; Durand, S.; van Diggelen, O.P.; Wevers, R.A.; Poorthuis, B.J.; Pshezhetsky, A.V.; Wijburg, F.A. Clinical and genetic spectrum of Sanfilippo type C (MPS IIIC) disease in The Netherlands. Mol. Genet. Metab. 2008, 93, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Valstar, M.J.; Marchal, J.P.; Grootenhuis, M.; Colland, V.; Wijburg, F.A. Cognitive development in patients with Mucopolysaccharidosis type III (Sanfilippo syndrome). Orphanet J. Rare Dis. 2011, 6, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, S.A.; Peracha, H.; Ballhausen, D.; Wiesbauer, A.; Rohrbach, M.; Gautschi, M.; Mason, R.W.; Giugliani, R.; Suzuki, Y.; Orii, K.E.; et al. Epidemiology of mucopolysaccharidoses. Mol. Genet. Metab. 2017, 121, 227–240. [Google Scholar] [CrossRef]
- Zelei, T.; Csetneki, K.; Voko, Z.; Siffel, C. Epidemiology of Sanfilippo syndrome: Results of a systematic literature review. Orphanet J. Rare Dis. 2018, 13, 53. [Google Scholar] [CrossRef] [Green Version]
- Scott, H.S.; Blanch, L.; Guo, X.H.; Freeman, C.; Orsborn, A.; Baker, E.; Sutherland, G.R.; Morris, C.P.; Hopwood, J.J. Cloning of the sulphamidase gene and identification of mutations in Sanfilippo A syndrome. Nat. Genet. 1995, 11, 465–467. [Google Scholar] [CrossRef]
- Zhao, H.G.; Li, H.H.; Bach, G.; Schmidtchen, A.; Neufeld, E.F. The molecular basis of Sanfilippo syndrome type B. Proc. Natl. Acad. Sci. USA 1996, 93, 6101–6105. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Zhang, H.; Zhang, S.; Bagshaw, R.D.; Tropak, M.B.; Callahan, J.W.; Mahuran, D.J. Identification of the gene encoding the enzyme deficient in mucopolysaccharidosis IIIC (Sanfilippo disease type C). Am. J. Hum. Genet. 2006, 79, 738–744. [Google Scholar] [CrossRef] [Green Version]
- Hrebicek, M.; Mrazova, L.; Seyrantepe, V.; Durand, S.; Roslin, N.M.; Noskova, L.; Hartmannova, H.; Ivanek, R.; Cizkova, A.; Poupetova, H.; et al. Mutations in TMEM76* cause mucopolysaccharidosis IIIC (Sanfilippo C syndrome). Am. J. Hum. Genet. 2006, 79, 807–819. [Google Scholar] [CrossRef] [Green Version]
- Durand, S.; Feldhammer, M.; Bonneil, E.; Thibault, P.; Pshezhetsky, A.V. Analysis of the biogenesis of heparan sulfate acetyl-CoA:alpha-glucosaminide N-acetyltransferase provides insights into the mechanism underlying its complete deficiency in mucopolysaccharidosis IIIC. J. Biol. Chem. 2010, 285, 31233–31242. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Tkachyova, I.; Sinha, A.; Rigat, B.; Mahuran, D. Characterization of the biosynthesis, processing and kinetic mechanism of action of the enzyme deficient in mucopolysaccharidosis IIIC. PLoS ONE 2011, 6, e24951. [Google Scholar] [CrossRef] [Green Version]
- Robertson, D.A.; Freeman, C.; Nelson, P.V.; Morris, C.P.; Hopwood, J.J. Human glucosamine-6-sulfatase cDNA reveals homology with steroid sulfatase. Biochem. Biophys. Res. Commun. 1988, 157, 218–224. [Google Scholar] [CrossRef]
- Li, J.P.; Kusche-Gullberg, M. Heparan Sulfate: Biosynthesis, Structure, and Function. Int. Rev. Cell Mol. Biol. 2016, 325, 215–273. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, F.; Sheng, J. “Coding” and “Decoding”: Hypothesis for the regulatory mechanism involved in heparan sulfate biosynthesis. Carbohydr. Res. 2016, 428, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Griffin, L.S.; Gloster, T.M. The Enzymatic Degradation of Heparan Sulfate. Protein Pept. Lett. 2017, 24, 710–722. [Google Scholar] [CrossRef] [PubMed]
- Pacifici, M. The pathogenic roles of heparan sulfate deficiency in hereditary multiple exostoses. Matrix Biol. 2018, 71–72, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Freeman, C.; Hopwood, J. Lysosomal degradation of heparin and heparan sulphate. Adv. Exp. Med. Biol. 1992, 313, 121–134. [Google Scholar] [CrossRef]
- Poulain, F.E. Analyzing the role of heparan sulfate proteoglycans in axon guidance in vivo in zebrafish. Methods Mol. Biol. 2015, 1229, 469–482. [Google Scholar] [CrossRef] [Green Version]
- Poulain, F.E.; Yost, H.J. Heparan sulfate proteoglycans: A sugar code for vertebrate development? Development 2015, 142, 3456–3467. [Google Scholar] [CrossRef] [Green Version]
- Long, K.R.; Huttner, W.B. How the extracellular matrix shapes neural development. Open Biol. 2019, 9, 180216. [Google Scholar] [CrossRef] [Green Version]
- Walkley, S.U. Secondary accumulation of gangliosides in lysosomal storage disorders. Semin. Cell Dev. Biol. 2004, 15, 433–444. [Google Scholar] [CrossRef]
- Bigger, B.W.; Begley, D.J.; Virgintino, D.; Pshezhetsky, A.V. Anatomical changes and pathophysiology of the brain in mucopolysaccharidosis disorders. Mol. Genet. Metab. 2018, 125, 322–331. [Google Scholar] [CrossRef]
- Fecarotta, S.; Gasperini, S.; Parenti, G. New treatments for the mucopolysaccharidoses: From pathophysiology to therapy. Ital. J. Pediatr. 2018, 44, 124. [Google Scholar] [CrossRef]
- Bruyere, J.; Roy, E.; Ausseil, J.; Lemonnier, T.; Teyre, G.; Bohl, D.; Etienne-Manneville, S.; Lortat-Jacob, H.; Heard, J.M.; Vitry, S. Heparan sulfate saccharides modify focal adhesions: Implication in mucopolysaccharidosis neuropathophysiology. J. Mol. Biol. 2015, 427, 775–791. [Google Scholar] [CrossRef]
- Martins, C.; Hulkova, H.; Dridi, L.; Dormoy-Raclet, V.; Grigoryeva, L.; Choi, Y.; Langford-Smith, A.; Wilkinson, F.L.; Ohmi, K.; DiCristo, G.; et al. Neuroinflammation, mitochondrial defects and neurodegeneration in mucopolysaccharidosis III type C mouse model. Brain 2015, 138, 336–355. [Google Scholar] [CrossRef] [Green Version]
- Winner, L.K.; Marshall, N.R.; Jolly, R.D.; Trim, P.J.; Duplock, S.K.; Snel, M.F.; Hemsley, K.M. Evaluation of Disease Lesions in the Developing Canine MPS IIIA Brain. JIMD Rep. 2019, 43, 91–101. [Google Scholar] [CrossRef]
- Wilkinson, F.L.; Holley, R.J.; Langford-Smith, K.J.; Badrinath, S.; Liao, A.; Langford-Smith, A.; Cooper, J.D.; Jones, S.A.; Wraith, J.E.; Wynn, R.F.; et al. Neuropathology in mouse models of mucopolysaccharidosis type I, IIIA and IIIB. PLoS ONE 2012, 7, e35787. [Google Scholar] [CrossRef]
- Parker, H.; Bigger, B.W. The role of innate immunity in mucopolysaccharide diseases. J. Neurochem. 2019, 148, 639–651. [Google Scholar] [CrossRef] [Green Version]
- Li, H.H.; Yu, W.H.; Rozengurt, N.; Zhao, H.Z.; Lyons, K.M.; Anagnostaras, S.; Fanselow, M.S.; Suzuki, K.; Vanier, M.T.; Neufeld, E.F. Mouse model of Sanfilippo syndrome type B produced by targeted disruption of the gene encoding alpha-N-acetylglucosaminidase. Proc. Natl. Acad. Sci. USA 1999, 96, 14505–14510. [Google Scholar] [CrossRef] [Green Version]
- Boustany, R.M. Lysosomal storage diseases—The horizon expands. Nat. Rev. Neurol. 2013, 9, 583–598. [Google Scholar] [CrossRef]
- Di Malta, C.; Fryer, J.D.; Settembre, C.; Ballabio, A. Astrocyte dysfunction triggers neurodegeneration in a lysosomal storage disorder. Proc. Natl. Acad. Sci. USA 2012, 109, E2334–E2342. [Google Scholar] [CrossRef] [Green Version]
- Scuderi, C.; Stecca, C.; Iacomino, A.; Steardo, L. Role of astrocytes in major neurological disorders: The evidence and implications. IUBMB Life 2013, 65, 957–961. [Google Scholar] [CrossRef]
- Settembre, C.; Fraldi, A.; Jahreiss, L.; Spampanato, C.; Venturi, C.; Medina, D.; de Pablo, R.; Tacchetti, C.; Rubinsztein, D.C.; Ballabio, A. A block of autophagy in lysosomal storage disorders. Hum. Mol. Genet. 2008, 17, 119–129. [Google Scholar] [CrossRef]
- Fischer, A.; Carmichael, K.P.; Munnell, J.F.; Jhabvala, P.; Thompson, J.N.; Matalon, R.; Jezyk, P.F.; Wang, P.; Giger, U. Sulfamidase deficiency in a family of Dachshunds: A canine model of mucopolysaccharidosis IIIA (Sanfilippo A). Pediatr. Res. 1998, 44, 74–82. [Google Scholar] [CrossRef] [Green Version]
- Jolly, R.D.; Allan, F.J.; Collett, M.G.; Rozaklis, T.; Muller, V.J.; Hopwood, J.J. Mucopolysaccharidosis IIIA (Sanfilippo syndrome) in a New Zealand Huntaway dog with ataxia. N. Z. Vet. J. 2000, 48, 144–148. [Google Scholar] [CrossRef]
- Bhaumik, M.; Muller, V.J.; Rozaklis, T.; Johnson, L.; Dobrenis, K.; Bhattacharyya, R.; Wurzelmann, S.; Finamore, P.; Hopwood, J.J.; Walkley, S.U.; et al. A mouse model for mucopolysaccharidosis type III A (Sanfilippo syndrome). Glycobiology 1999, 9, 1389–1396. [Google Scholar] [CrossRef]
- Crawley, A.C.; Gliddon, B.L.; Auclair, D.; Brodie, S.L.; Hirte, C.; King, B.M.; Fuller, M.; Hemsley, K.M.; Hopwood, J.J. Characterization of a C57BL/6 congenic mouse strain of mucopolysaccharidosis type IIIA. Brain Res. 2006, 1104, 1–17. [Google Scholar] [CrossRef]
- Ellinwood, N.M.; Wang, P.; Skeen, T.; Sharp, N.J.; Cesta, M.; Decker, S.; Edwards, N.J.; Bublot, I.; Thompson, J.N.; Bush, W.; et al. A model of mucopolysaccharidosis IIIB (Sanfilippo syndrome type IIIB): N-acetyl-alpha-D-glucosaminidase deficiency in Schipperke dogs. J. Inherit. Metab. Dis. 2003, 26, 489–504. [Google Scholar] [CrossRef]
- Aronovich, E.L.; Johnston, J.M.; Wang, P.; Giger, U.; Whitley, C.B. Molecular basis of mucopolysaccharidosis type IIIB in emu (Dromaius novaehollandiae): An avian model of Sanfilippo syndrome type B. Genomics 2001, 74, 299–305. [Google Scholar] [CrossRef]
- Karageorgos, L.; Hill, B.; Bawden, M.J.; Hopwood, J.J. Bovine mucopolysaccharidosis type IIIB. J. Inherit. Metab. Dis. 2007, 30, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Marco, S.; Pujol, A.; Roca, C.; Motas, S.; Ribera, A.; Garcia, M.; Molas, M.; Villacampa, P.; Melia, C.S.; Sanchez, V.; et al. Progressive neurologic and somatic disease in a novel mouse model of human mucopolysaccharidosis type IIIC. Dis. Model Mech. 2016, 9, 999–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, J.N.; Jones, M.Z.; Dawson, G.; Huffman, P.S. N-acetylglucosamine 6-sulphatase deficiency in a Nubian goat: A model of Sanfilippo syndrome type D (mucopolysaccharidosis IIID). J. Inherit. Metab. Dis. 1992, 15, 760–768. [Google Scholar] [CrossRef]
- Roca, C.; Motas, S.; Marco, S.; Ribera, A.; Sanchez, V.; Sanchez, X.; Bertolin, J.; Leon, X.; Perez, J.; Garcia, M.; et al. Disease correction by AAV-mediated gene therapy in a new mouse model of mucopolysaccharidosis type IIID. Hum. Mol. Genet. 2017, 26, 1535–1551. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Lemonnier, T.; Blanchard, S.; Toli, D.; Roy, E.; Bigou, S.; Froissart, R.; Rouvet, I.; Vitry, S.; Heard, J.M.; Bohl, D. Modeling neuronal defects associated with a lysosomal disorder using patient-derived induced pluripotent stem cells. Hum. Mol. Genet. 2011, 20, 3653–3666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canals, I.; Soriano, J.; Orlandi, J.G.; Torrent, R.; Richaud-Patin, Y.; Jimenez-Delgado, S.; Merlin, S.; Follenzi, A.; Consiglio, A.; Vilageliu, L.; et al. Activity and High-Order Effective Connectivity Alterations in Sanfilippo C Patient-Specific Neuronal Networks. Stem Cell Rep. 2015, 5, 546–557. [Google Scholar] [CrossRef] [Green Version]
- Vallejo, S.; Fleischer, A.; Martin, J.M.; Sanchez, A.; Palomino, E.; Bachiller, D. Generation of two induced pluripotent stem cells lines from Mucopolysaccharydosis IIIA patient: IMEDEAi004-A and IMEDEAi004-B. Stem Cell Res. 2018, 32, 110–114. [Google Scholar] [CrossRef]
- Vallejo-Diez, S.; Fleischer, A.; Martin-Fernandez, J.M.; Sanchez-Gilabert, A.; Bachiller, D. Generation of two induced pluripotent stem cells lines from a Mucopolysaccharydosis IIIB (MPSIIIB) patient. Stem Cell Res. 2018, 33, 180–184. [Google Scholar] [CrossRef]
- Huang, W.; Xu, M.; Li, R.; Baskfield, A.; Kouznetsova, J.; Beers, J.; Zou, J.; Liu, C.; Zheng, W. An induced pluripotent stem cell line (TRNDi006-A) from a MPS IIIB patient carrying homozygous mutation of p.Glu153Lys in the NAGLU gene. Stem Cell Res. 2019, 37, 101427. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Li, X.F.; Zhou, Y.W.; Cai, P.F.; Fu, W.C.; Wang, J.H.; Chen, J.Y.; Yang, Q.N. CRISPR/Cas9 facilitates genomic editing for large-scale functional studies in pluripotent stem cell cultures. Hum. Genet. 2019, 138, 1217–1225. [Google Scholar] [CrossRef]
- Beneto, N.; Cozar, M.; Garcia-Morant, M.; Creus-Bachiller, E.; Vilageliu, L.; Grinberg, D.; Canals, I. Generation of two compound heterozygous HGSNAT-mutated lines from healthy induced pluripotent stem cells using CRISPR/Cas9 to model Sanfilippo C syndrome. Stem Cell Res. 2019, 41, 101616. [Google Scholar] [CrossRef] [PubMed]
- Beneto, N.; Cozar, M.; Castilla-Vallmanya, L.; Zetterdahl, O.G.; Sacultanu, M.; Segur-Bailach, E.; Garcia-Morant, M.; Ribes, A.; Ahlenius, H.; Grinberg, D.; et al. Neuronal and Astrocytic Differentiation from Sanfilippo C Syndrome iPSCs for Disease Modeling and Drug Development. J. Clin. Med. 2020, 9, 644. [Google Scholar] [CrossRef] [Green Version]
- Beneto, N.; Cozar, M.; Gort, L.; Pacheco, L.; Vilageliu, L.; Grinberg, D.; Canals, I. Generation of two NAGLU-mutated homozygous cell lines from healthy induced pluripotent stem cells using CRISPR/Cas9 to model Sanfilippo B syndrome. Stem Cell Res. 2019, 42, 101668. [Google Scholar] [CrossRef]
- Matalonga, L.; Arias, A.; Coll, M.J.; Garcia-Villoria, J.; Gort, L.; Ribes, A. Treatment effect of coenzyme Q(10) and an antioxidant cocktail in fibroblasts of patients with Sanfilippo disease. J. Inherit. Metab. Dis. 2014, 37, 439–446. [Google Scholar] [CrossRef]
- Lotfi, P.; Tse, D.Y.; Di Ronza, A.; Seymour, M.L.; Martano, G.; Cooper, J.D.; Pereira, F.A.; Passafaro, M.; Wu, S.M.; Sardiello, M. Trehalose reduces retinal degeneration, neuroinflammation and storage burden caused by a lysosomal hydrolase deficiency. Autophagy 2018, 14, 1419–1434. [Google Scholar] [CrossRef] [Green Version]
- Bajaj, L.; Lotfi, P.; Pal, R.; Ronza, A.D.; Sharma, J.; Sardiello, M. Lysosome biogenesis in health and disease. J. Neurochem. 2019, 148, 573–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napolitano, G.; Ballabio, A. TFEB at a glance. J. Cell Sci. 2016, 129, 2475–2481. [Google Scholar] [CrossRef] [Green Version]
- Matos, L.; Canals, I.; Dridi, L.; Choi, Y.; Prata, M.J.; Jordan, P.; Desviat, L.R.; Perez, B.; Pshezhetsky, A.V.; Grinberg, D.; et al. Therapeutic strategies based on modified U1 snRNAs and chaperones for Sanfilippo C splicing mutations. Orphanet J. Rare Dis. 2014, 9, 180. [Google Scholar] [CrossRef] [Green Version]
- Tomanin, R.; Zanetti, A.; Zaccariotto, E.; D’Avanzo, F.; Bellettato, C.M.; Scarpa, M. Gene therapy approaches for lysosomal storage disorders, a good model for the treatment of mendelian diseases. Acta Paediatr. 2012, 101, 692–701. [Google Scholar] [CrossRef] [PubMed]
- Parenti, G.; Andria, G.; Ballabio, A. Lysosomal storage diseases: From pathophysiology to therapy. Annu. Rev. Med. 2015, 66, 471–486. [Google Scholar] [CrossRef] [PubMed]
- Kishnani, P.S.; Dickson, P.I.; Muldowney, L.; Lee, J.J.; Rosenberg, A.; Abichandani, R.; Bluestone, J.A.; Burton, B.K.; Dewey, M.; Freitas, A.; et al. Immune response to enzyme replacement therapies in lysosomal storage diseases and the role of immune tolerance induction. Mol. Genet. Metab. 2016, 117, 66–83. [Google Scholar] [CrossRef] [Green Version]
- Grover, A.; Crippen-Harmon, D.; Nave, L.; Vincelette, J.; Wait, J.C.M.; Melton, A.C.; Lawrence, R.; Brown, J.R.; Webster, K.A.; Yip, B.K.; et al. Translational studies of intravenous and intracerebroventricular routes of administration for CNS cellular biodistribution for BMN 250, an enzyme replacement therapy for the treatment of Sanfilippo type B. Drug Deliv. Transl. Res. 2020, 10, 425–439. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.A.; Breen, C.; Heap, F.; Rust, S.; de Ruijter, J.; Tump, E.; Marchal, J.P.; Pan, L.; Qiu, Y.; Chung, J.K.; et al. A phase 1/2 study of intrathecal heparan-N-sulfatase in patients with mucopolysaccharidosis IIIA. Mol. Genet. Metab. 2016, 118, 198–205. [Google Scholar] [CrossRef]
- Wijburg, F.A.; Whitley, C.B.; Muenzer, J.; Gasperini, S.; Del Toro, M.; Muschol, N.; Cleary, M.; Sevin, C.; Shapiro, E.; Bhargava, P.; et al. Intrathecal heparan-N-sulfatase in patients with Sanfilippo syndrome type A: A phase IIb randomized trial. Mol. Genet. Metab. 2019, 126, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Muschol, N.; Cleary, M.; Couce, M.L.; Shaywitz, A.J.; Cahan, H.; Grover, A.; Maricich, S.M.; Melton, A.; Smith, L.; Lopez, M.J.D. ICV-administered BMN 250 (NAGLU-IGF2) is well tolerated and reduces heparan sulfate accumulation in the CNS of subjects with Sanfilippo syndrome type B (MPS IIIB). Mol. Genet. Metab. 2018, 123, S102. [Google Scholar] [CrossRef]
- Whitley, C.B.; Vijay, S.; Yao, B.; Pineda, M.; Parker, G.J.M.; Rojas-Caro, S.; Zhang, X.; Dai, Y.; Cinar, A.; Bubb, G.; et al. Final results of the phase 1/2, open-label clinical study of intravenous recombinant human N-acetyl-alpha-d-glucosaminidase (SBC-103) in children with mucopolysaccharidosis IIIB. Mol. Genet. Metab. 2019, 126, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Sawamoto, K.; Stapleton, M.; Almeciga-Diaz, C.J.; Espejo-Mojica, A.J.; Losada, J.C.; Suarez, D.A.; Tomatsu, S. Therapeutic Options for Mucopolysaccharidoses: Current and Emerging Treatments. Drugs 2019, 79, 1103–1134. [Google Scholar] [CrossRef]
- Venier, R.E.; Igdoura, S.A. Miglustat as a therapeutic agent: Prospects and caveats. J. Med. Genet. 2012, 49, 591–597. [Google Scholar] [CrossRef]
- Pineda, M.; Walterfang, M.; Patterson, M.C. Miglustat in Niemann-Pick disease type C patients: A review. Orphanet J. Rare Dis. 2018, 13, 140. [Google Scholar] [CrossRef]
- Jakobkiewicz-Banecka, J.; Piotrowska, E.; Narajczyk, M.; Baranska, S.; Wegrzyn, G. Genistein-mediated inhibition of glycosaminoglycan synthesis, which corrects storage in cells of patients suffering from mucopolysaccharidoses, acts by influencing an epidermal growth factor-dependent pathway. J. Biomed. Sci. 2009, 16, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malinowska, M.; Wilkinson, F.L.; Langford-Smith, K.J.; Langford-Smith, A.; Brown, J.R.; Crawford, B.E.; Vanier, M.T.; Grynkiewicz, G.; Wynn, R.F.; Wraith, J.E.; et al. Genistein improves neuropathology and corrects behaviour in a mouse model of neurodegenerative metabolic disease. PLoS ONE 2010, 5, e14192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgadillo, V.; O’Callaghan Mdel, M.; Artuch, R.; Montero, R.; Pineda, M. Genistein supplementation in patients affected by Sanfilippo disease. J. Inherit. Metab. Dis. 2011, 34, 1039–1044. [Google Scholar] [CrossRef]
- de Ruijter, J.; Valstar, M.J.; Narajczyk, M.; Wegrzyn, G.; Kulik, W.; Ijlst, L.; Wagemans, T.; van der Wal, W.M.; Wijburg, F.A. Genistein in Sanfilippo disease: A randomized controlled crossover trial. Ann. Neurol. 2012, 71, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Dziedzic, D.; Wegrzyn, G.; Jakobkiewicz-Banecka, J. Impairment of glycosaminoglycan synthesis in mucopolysaccharidosis type IIIA cells by using siRNA: A potential therapeutic approach for Sanfilippo disease. Eur. J. Hum. Genet. 2010, 18, 200–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaidonis, X.; Liaw, W.C.; Roberts, A.D.; Ly, M.; Anson, D.; Byers, S. Gene silencing of EXTL2 and EXTL3 as a substrate deprivation therapy for heparan sulphate storing mucopolysaccharidoses. Eur. J. Hum. Genet. 2010, 18, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Canals, I.; Beneto, N.; Cozar, M.; Vilageliu, L.; Grinberg, D. EXTL2 and EXTL3 inhibition with siRNAs as a promising substrate reduction therapy for Sanfilippo C syndrome. Sci. Rep. 2015, 5, 13654. [Google Scholar] [CrossRef]
- Suzuki, Y. Chaperone therapy for molecular pathology in lysosomal diseases. Brain Dev. 2020. [Google Scholar] [CrossRef]
- Losada Diaz, J.C.; Cepeda Del Castillo, J.; Rodriguez-Lopez, E.A.; Almeciga-Diaz, C.J. Advances in the Development of Pharmacological Chaperones for the Mucopolysaccharidoses. Int. J. Mol. Sci. 2019, 21, 232. [Google Scholar] [CrossRef] [Green Version]
- Parenti, G.; Andria, G.; Valenzano, K.J. Pharmacological Chaperone Therapy: Preclinical Development, Clinical Translation, and Prospects for the Treatment of Lysosomal Storage Disorders. Mol. Ther. 2015, 23, 1138–1148. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, Y. Emerging novel concept of chaperone therapies for protein misfolding diseases. Proc. Jpn. Acad. Ser. B 2014, 90, 145–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldhammer, M.; Durand, S.; Pshezhetsky, A.V. Protein misfolding as an underlying molecular defect in mucopolysaccharidosis III type C. PLoS ONE 2009, 4, e7434. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.A.; Hannouche, H.; Rozaklis, T.; Hassiotis, S.; Hopwood, J.J.; Hemsley, K.M. Allogeneic stem cell transplantation does not improve neurological deficits in mucopolysaccharidosis type IIIA mice. Exp. Neurol. 2010, 225, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.A.; Shamsani, N.J.; Winner, L.K.; Hassiotis, S.; King, B.M.; Hopwood, J.J.; Hemsley, K.M. Neonatal Bone Marrow Transplantation in MPS IIIA Mice. JIMD Rep. 2013, 8, 121–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krivit, W.; Sung, J.H.; Shapiro, E.G.; Lockman, L.A. Microglia: The effector cell for reconstitution of the central nervous system following bone marrow transplantation for lysosomal and peroxisomal storage diseases. Cell Transpl. 1995, 4, 385–392. [Google Scholar] [CrossRef]
- Tan, E.Y.; Boelens, J.J.; Jones, S.A.; Wynn, R.F. Hematopoietic Stem Cell Transplantation in Inborn Errors of Metabolism. Front. Pediatr. 2019, 7, 433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langford-Smith, A.; Wilkinson, F.L.; Langford-Smith, K.J.; Holley, R.J.; Sergijenko, A.; Howe, S.J.; Bennett, W.R.; Jones, S.A.; Wraith, J.E.; Merry, C.L.R.; et al. Hematopoietic Stem Cell and Gene Therapy Corrects Primary Neuropathology and Behavior in Mucopolysaccharidosis IIIA Mice. Mol. Ther. 2012, 20, 1610–1621. [Google Scholar] [CrossRef] [Green Version]
- Holley, R.J.; Ellison, S.M.; Fil, D.; O’Leary, C.; McDermott, J.; Senthivel, N.; Langford-Smith, A.W.W.; Wilkinson, F.L.; D’Souza, Z.; Parker, H.; et al. Macrophage enzyme and reduced inflammation drive brain correction of mucopolysaccharidosis IIIB by stem cell gene therapy. Brain 2018, 141, 99–116. [Google Scholar] [CrossRef] [Green Version]
- Sergijenko, A.; Langford-Smith, A.; Liao, A.Y.; Pickford, C.E.; McDermott, J.; Nowinski, G.; Langford-Smith, K.J.; Merry, C.L.; Jones, S.A.; Wraith, J.E.; et al. Myeloid/Microglial driven autologous hematopoietic stem cell gene therapy corrects a neuronopathic lysosomal disease. Mol. Ther. 2013, 21, 1938–1949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellison, S.M.; Liao, A.; Wood, S.; Taylor, J.; Youshani, A.S.; Rowlston, S.; Parker, H.; Armant, M.; Biffi, A.; Chan, L.; et al. Pre-clinical Safety and Efficacy of Lentiviral Vector-Mediated Ex Vivo Stem Cell Gene Therapy for the Treatment of Mucopolysaccharidosis IIIA. Mol. Ther. Methods Clin. Dev. 2019, 13, 399–413. [Google Scholar] [CrossRef] [Green Version]
- Willing, A.E.; Garbuzova-Davis, S.N.; Zayko, O.; Derasari, H.M.; Rawls, A.E.; James, C.R.; Mervis, R.F.; Sanberg, C.D.; Kuzmin-Nichols, N.; Sanberg, P.R. Repeated administrations of human umbilical cord blood cells improve disease outcomes in a mouse model of Sanfilippo syndrome type III B. Cell Transpl. 2014, 23, 1613–1630. [Google Scholar] [CrossRef] [PubMed]
- Welling, L.; Marchal, J.P.; van Hasselt, P.; van der Ploeg, A.T.; Wijburg, F.A.; Boelens, J.J. Early Umbilical Cord Blood-Derived Stem Cell Transplantation Does Not Prevent Neurological Deterioration in Mucopolysaccharidosis Type III. JIMD Rep. 2015, 18, 63–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebrahimi, T.; Abasi, M.; Seifar, F.; Eyvazi, S.; Hejazi, M.S.; Tarhriz, V.; Montazersaheb, S. Transplantation of Stem Cells as a Potential Therapeutic Strategy in Neurodegenerative Disorders. Curr. Stem Cell Res. Ther. 2020. [Google Scholar] [CrossRef] [PubMed]
- Griffin, T.A.; Anderson, H.C.; Wolfe, J.H. Ex vivo gene therapy using patient iPSC-derived NSCs reverses pathology in the brain of a homologous mouse model. Stem Cell Rep. 2015, 4, 835–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, D.; Pearse, Y.; Kan, S.H.; Le, S.Q.; Sanghez, V.; Cooper, J.D.; Dickson, P.I.; Iacovino, M. Genetically Corrected iPSC-Derived Neural Stem Cell Grafts Deliver Enzyme Replacement to Affect CNS Disease in Sanfilippo B Mice. Mol. Ther. Methods Clin. Dev. 2018, 10, 113–127. [Google Scholar] [CrossRef] [Green Version]
- Robinson, A.J.; Zhao, G.; Rathjen, J.; Rathjen, P.D.; Hutchinson, R.G.; Eyre, H.J.; Hemsley, K.M.; Hopwood, J.J. Embryonic stem cell-derived glial precursors as a vehicle for sulfamidase production in the MPS-IIIA mouse brain. Cell Transpl. 2010, 19, 985–998. [Google Scholar] [CrossRef]
- Izrael, M.; Slutsky, S.G.; Revel, M. Rising Stars: Astrocytes as a Therapeutic Target for ALS Disease. Front. Neurosci. 2020, 14, 824. [Google Scholar] [CrossRef]
- Quiviger, M.; Arfi, A.; Mansard, D.; Delacotte, L.; Pastor, M.; Scherman, D.; Marie, C. High and prolonged sulfamidase secretion by the liver of MPS-IIIA mice following hydrodynamic tail vein delivery of antibiotic-free pFAR4 plasmid vector. Gene Ther. 2014, 21, 1001–1007. [Google Scholar] [CrossRef]
- Johnston, S.; Parylak, S.; Kim, S.; Mac, N.; Lim, C.; Gallina, I.; Bloyd, C.; Newberry, A.; Saavedra, C.; Novák, O.; et al. AAV Ablates Neurogenesis in the Adult Murine Hippocampus. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Fraldi, A.; Hemsley, K.; Crawley, A.; Lombardi, A.; Lau, A.; Sutherland, L.; Auricchio, A.; Ballabio, A.; Hopwood, J.J. Functional correction of CNS lesions in an MPS-IIIA mouse model by intracerebral AAV-mediated delivery of sulfamidase and SUMF1 genes. Hum. Mol. Genet. 2007, 16, 2693–2702. [Google Scholar] [CrossRef] [Green Version]
- Tardieu, M.; Zerah, M.; Husson, B.; de Bournonville, S.; Deiva, K.; Adamsbaum, C.; Vincent, F.; Hocquemiller, M.; Broissand, C.; Furlan, V.; et al. Intracerebral administration of adeno-associated viral vector serotype rh.10 carrying human SGSH and SUMF1 cDNAs in children with mucopolysaccharidosis type IIIA disease: Results of a phase I/II trial. Hum. Gene Ther. 2014, 25, 506–516. [Google Scholar] [CrossRef]
- Tardieu, M.; Zerah, M.; Gougeon, M.L.; Ausseil, J.; de Bournonville, S.; Husson, B.; Zafeiriou, D.; Parenti, G.; Bourget, P.; Poirier, B.; et al. Intracerebral gene therapy in children with mucopolysaccharidosis type IIIB syndrome: An uncontrolled phase 1/2 clinical trial. Lancet Neurol. 2017, 16, 712–720. [Google Scholar] [CrossRef]
- Winner, L.K.; Beard, H.; Hassiotis, S.; Lau, A.A.; Luck, A.J.; Hopwood, J.J.; Hemsley, K.M. A Preclinical Study Evaluating AAVrh10-Based Gene Therapy for Sanfilippo Syndrome. Hum. Gene Ther. 2016, 27, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Hocquemiller, M.; Hemsley, K.M.; Douglass, M.L.; Tamang, S.J.; Neumann, D.; King, B.M.; Beard, H.; Trim, P.J.; Winner, L.K.; Lau, A.A.; et al. AAVrh10 Vector Corrects Disease Pathology in MPS IIIA Mice and Achieves Widespread Distribution of SGSH in Large Animal Brains. Mol. Ther. Methods Clin. Dev. 2020, 17, 174–187. [Google Scholar] [CrossRef] [Green Version]
- Gilkes, J.A.; Bloom, M.D.; Heldermon, C.D. Preferred transduction with AAV8 and AAV9 via thalamic administration in the MPS IIIB model: A comparison of four rAAV serotypes. Mol. Genet. Metab. Rep. 2016, 6, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Ruzo, A.; Garcia, M.; Ribera, A.; Villacampa, P.; Haurigot, V.; Marco, S.; Ayuso, E.; Anguela, X.M.; Roca, C.; Agudo, J.; et al. Liver production of sulfamidase reverses peripheral and ameliorates CNS pathology in mucopolysaccharidosis IIIA mice. Mol. Ther. 2012, 20, 254–266. [Google Scholar] [CrossRef]
- Sorrentino, N.C.; D’Orsi, L.; Sambri, I.; Nusco, E.; Monaco, C.; Spampanato, C.; Polishchuk, E.; Saccone, P.; De Leonibus, E.; Ballabio, A.; et al. A highly secreted sulphamidase engineered to cross the blood-brain barrier corrects brain lesions of mice with mucopolysaccharidoses type IIIA. EMBO Mol. Med. 2013, 5, 675–690. [Google Scholar] [CrossRef]
- Murrey, D.A.; Naughton, B.J.; Duncan, F.J.; Meadows, A.S.; Ware, T.A.; Campbell, K.J.; Bremer, W.G.; Walker, C.M.; Goodchild, L.; Bolon, B.; et al. Feasibility and safety of systemic rAAV9-hNAGLU delivery for treating mucopolysaccharidosis IIIB: Toxicology, biodistribution, and immunological assessments in primates. Hum. Gene Ther. Clin. Dev. 2014, 25, 72–84. [Google Scholar] [CrossRef] [Green Version]
- Haurigot, V.; Marco, S.; Ribera, A.; Garcia, M.; Ruzo, A.; Villacampa, P.; Ayuso, E.; Anor, S.; Andaluz, A.; Pineda, M.; et al. Whole body correction of mucopolysaccharidosis IIIA by intracerebrospinal fluid gene therapy. J. Clin. Investig. 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruzo, A.; Marco, S.; Garcia, M.; Villacampa, P.; Ribera, A.; Ayuso, E.; Maggioni, L.; Mingozzi, F.; Haurigot, V.; Bosch, F. Correction of pathological accumulation of glycosaminoglycans in central nervous system and peripheral tissues of MPSIIIA mice through systemic AAV9 gene transfer. Hum. Gene Ther. 2012, 23, 1237–1246. [Google Scholar] [CrossRef]
- Ribera, A.; Haurigot, V.; Garcia, M.; Marco, S.; Motas, S.; Villacampa, P.; Maggioni, L.; Leon, X.; Molas, M.; Sanchez, V.; et al. Biochemical, histological and functional correction of mucopolysaccharidosis type IIIB by intra-cerebrospinal fluid gene therapy. Hum. Mol. Genet. 2015, 24, 2078–2095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, H.; McCarty, D.M. Crossing the blood-brain-barrier with viral vectors. Curr. Opin. Virol. 2016, 21, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Tordo, J.; O’Leary, C.; Antunes, A.; Palomar, N.; Aldrin-Kirk, P.; Basche, M.; Bennett, A.; D’Souza, Z.; Gleitz, H.; Godwin, A.; et al. A novel adeno-associated virus capsid with enhanced neurotropism corrects a lysosomal transmembrane enzyme deficiency. Brain 2018, 141, 2014–2031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flitsch, L.J.; Laupman, K.E.; Brustle, O. Transcription Factor-Based Fate Specification and Forward Programming for Neural Regeneration. Front. Cell. Neurosci. 2020, 14, 121. [Google Scholar] [CrossRef]
- Tao, Y.; Zhang, S.C. Neural Subtype Specification from Human Pluripotent Stem Cells. Cell Stem Cell 2016, 19, 573–586. [Google Scholar] [CrossRef] [Green Version]
- Velasco, S.; Paulsen, B.; Arlotta, P. 3D Brain Organoids: Studying Brain Development and Disease Outside the Embryo. Annu. Rev. Neurosci. 2020, 43, 375–389. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
| Total Mutations | Missense/Nonsense | Small Deletions | Small Insertions | Small Indels | Splicing | Gross Deletions | Gross Insertions and Duplications | Complex Rearrangements | |
|---|---|---|---|---|---|---|---|---|---|
| A (SGSH) | 155 | 118 | 20 | 9 | 1 | 3 | 3 | 1 | 0 |
| B (NAGLU) | 229 | 167 | 29 | 16 | 1 | 8 | 4 | 4 | 0 |
| C (HGSNAT) | 77 | 43 | 6 | 6 | 1 | 15 | 4 | 1 | 1 |
| D (GNS) | 25 | 7 | 5 | 4 | 1 | 4 | 2 | 0 | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benetó, N.; Vilageliu, L.; Grinberg, D.; Canals, I. Sanfilippo Syndrome: Molecular Basis, Disease Models and Therapeutic Approaches. Int. J. Mol. Sci. 2020, 21, 7819. https://doi.org/10.3390/ijms21217819
Benetó N, Vilageliu L, Grinberg D, Canals I. Sanfilippo Syndrome: Molecular Basis, Disease Models and Therapeutic Approaches. International Journal of Molecular Sciences. 2020; 21(21):7819. https://doi.org/10.3390/ijms21217819
Chicago/Turabian StyleBenetó, Noelia, Lluïsa Vilageliu, Daniel Grinberg, and Isaac Canals. 2020. "Sanfilippo Syndrome: Molecular Basis, Disease Models and Therapeutic Approaches" International Journal of Molecular Sciences 21, no. 21: 7819. https://doi.org/10.3390/ijms21217819