

NeoCoV Is Closer to MERS-CoV than SARS-CoV
- PMID: 32595278
- PMCID: PMC7298434
- DOI: 10.1177/1178633720930711
NeoCoV Is Closer to MERS-CoV than SARS-CoV
Abstract
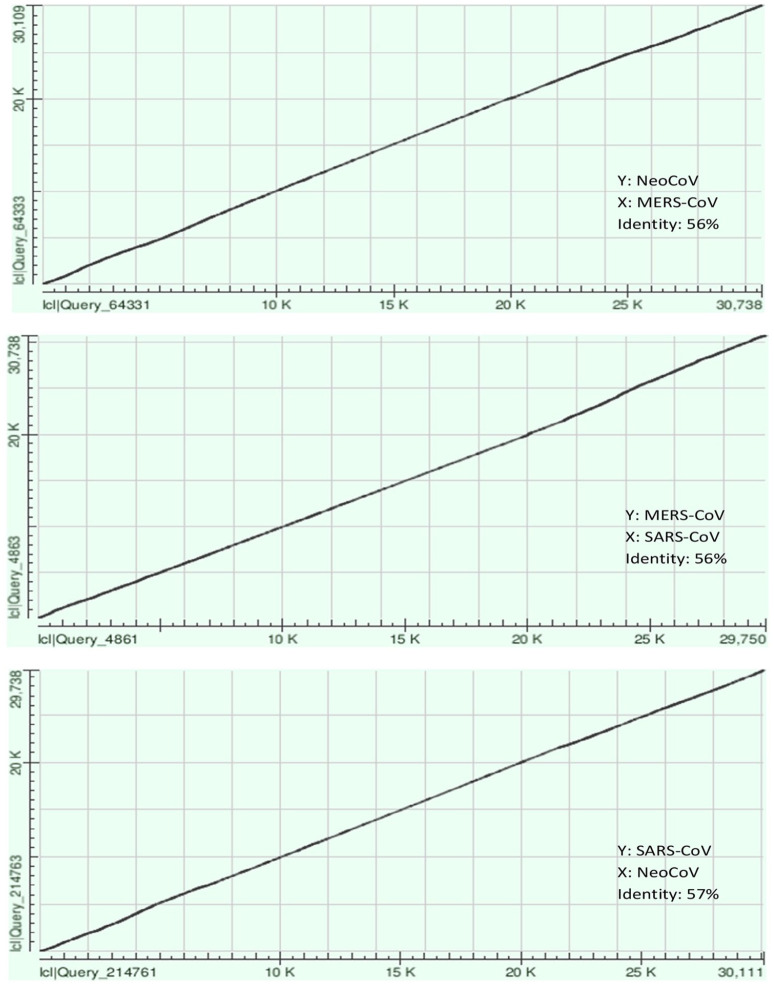
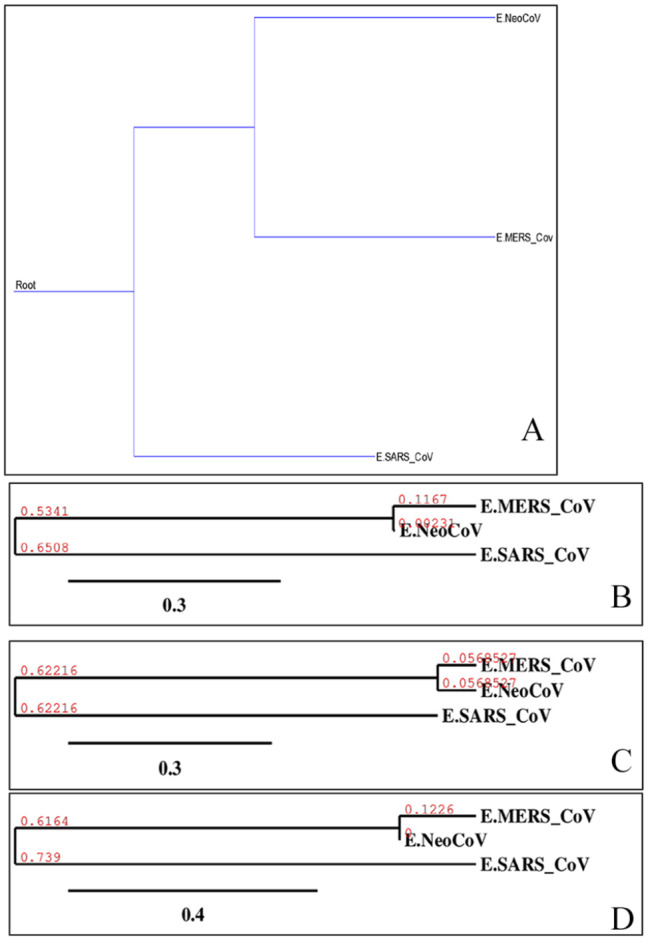
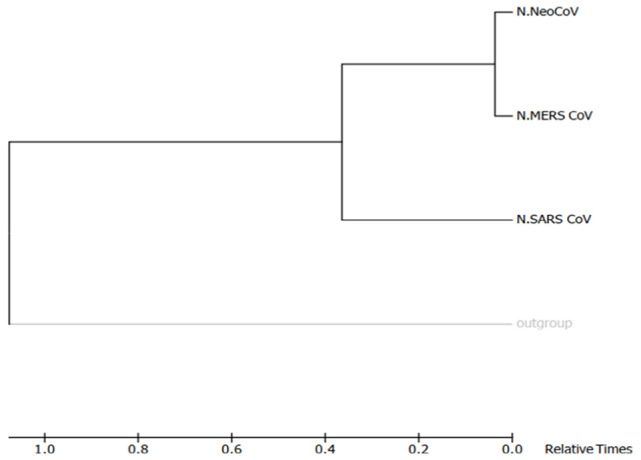
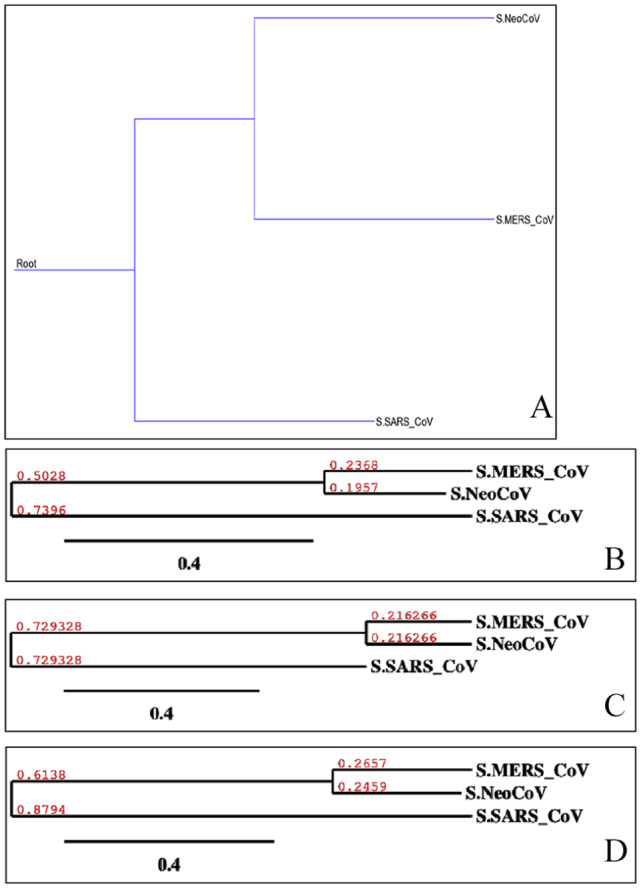
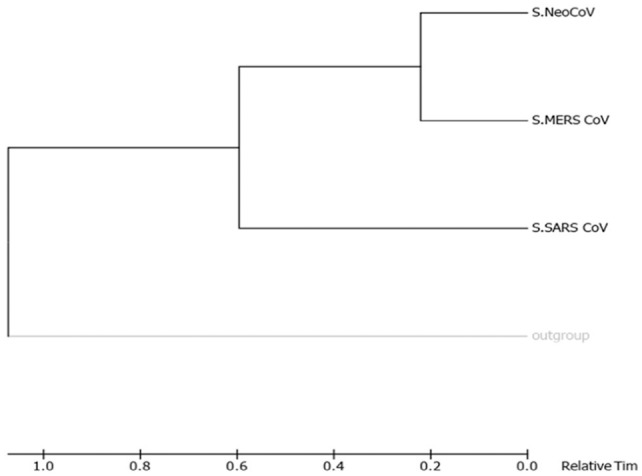
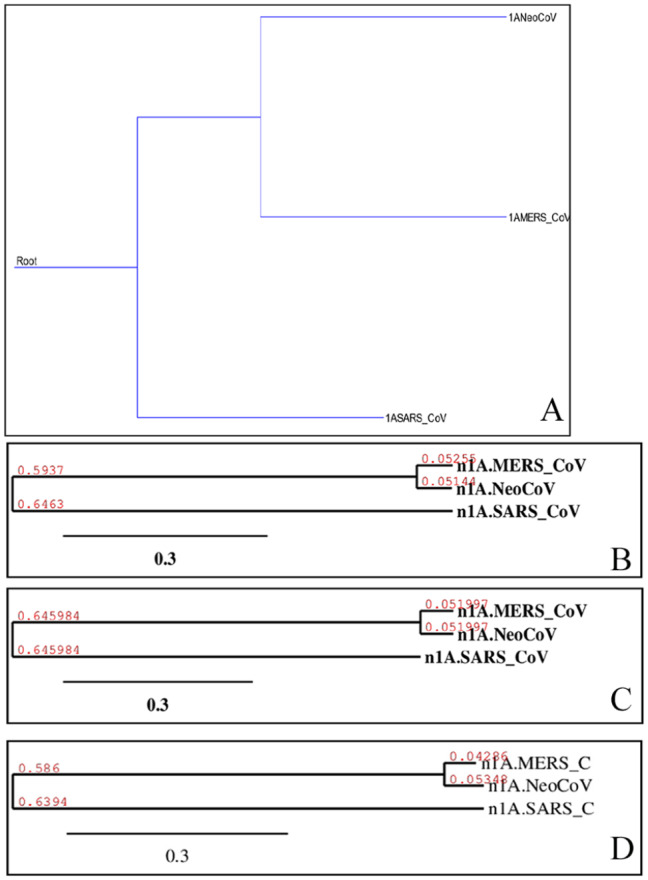
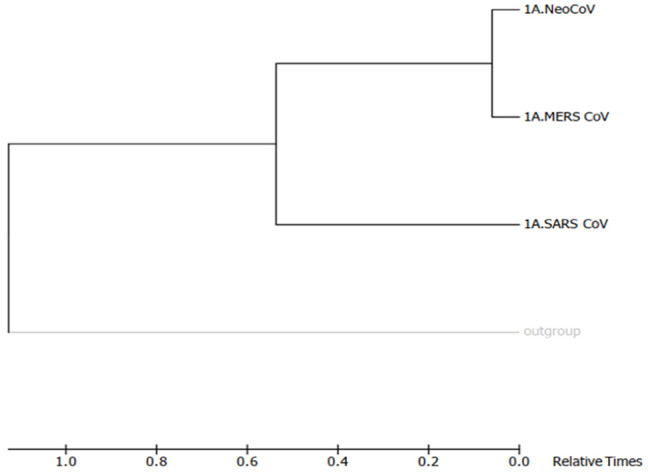
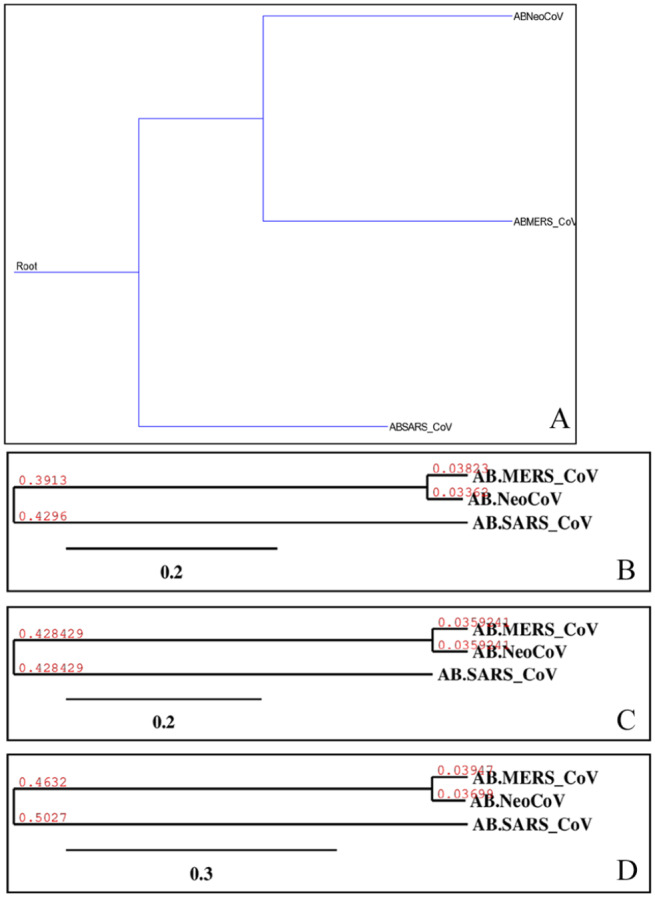
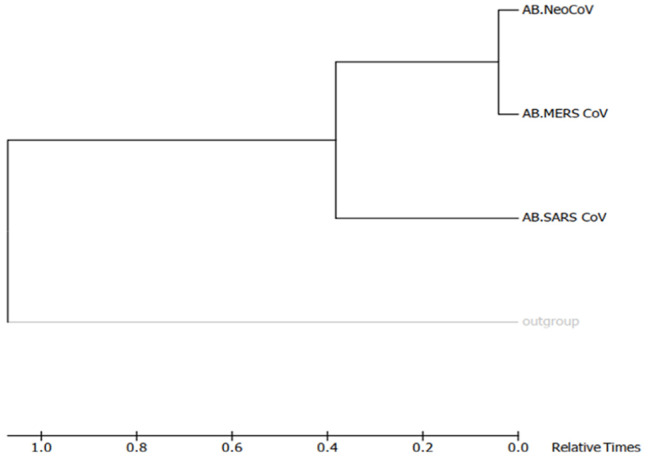
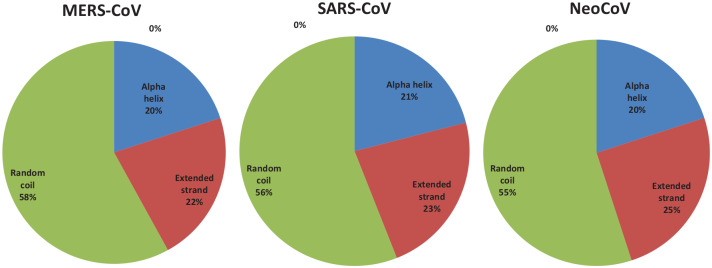
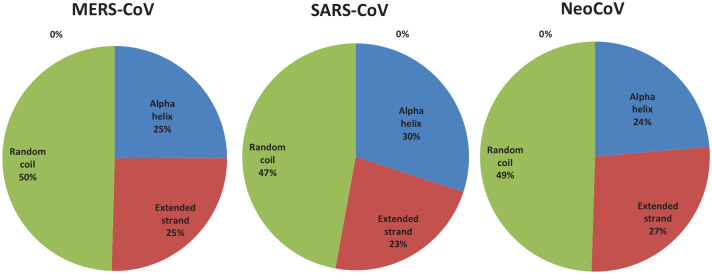
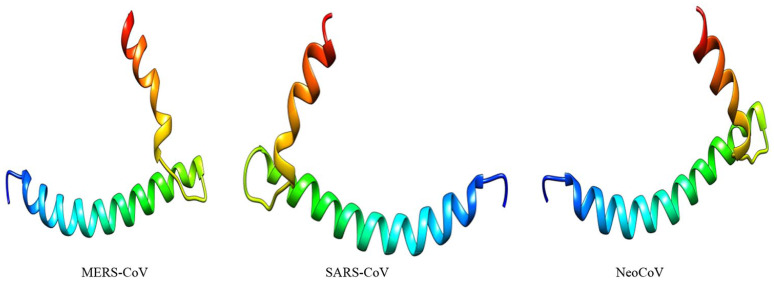
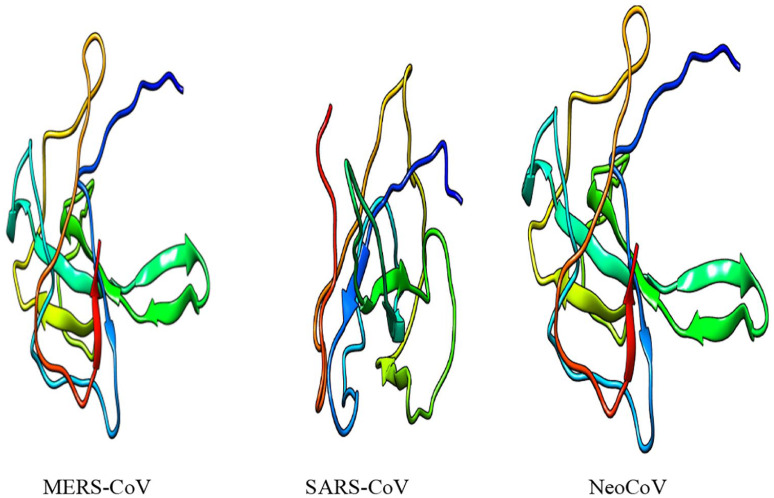
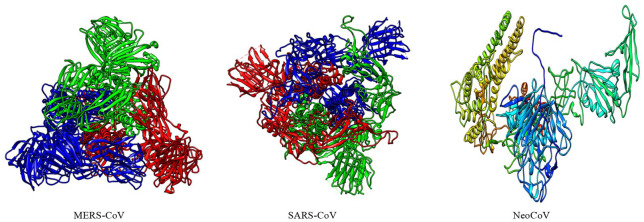
Recently, Coronavirus has been given considerable attention from the biomedical community based on the emergence and isolation of a deadly coronavirus infecting human. To understand the behavior of the newly emerging MERS-CoV requires knowledge at different levels (epidemiologic, antigenic, and pathogenic), and this knowledge can be generated from the most related viruses. In this study, we aimed to compare between 3 species of Coronavirus, namely Middle East Respiratory Syndrome (MERS-CoV), Severe Acute Respiratory Syndrome (SARS-CoV), and NeoCoV regarding whole genomes and 6 similar proteins (E, M, N, S, ORF1a, and ORF1ab) using different bioinformatics tools to provide a better understanding of the relationship between the 3 viruses at the nucleotide and amino acids levels. All sequences have been retrieved from National Center for Biotechnology Information (NCBI). Regards to target genomes' phylogenetic analysis showed that MERS and SARS-CoVs were closer to each other compared with NeoCoV, and the last has the longest relative time. We found that all phylogenetic methods in addition to all parameters (physical and chemical properties of amino acids such as the number of amino acid, molecular weight, atomic composition, theoretical pI, and structural formula) indicated that NeoCoV proteins were the most related to MERS-CoV one. All phylogenetic trees (by both maximum-likelihood and neighbor-joining methods) indicated that NeoCoV proteins have less evolutionary changes except for ORF1a by just maximum-likelihood method. Our results indicated high similarity between viral structural proteins which are responsible for viral infectivity; therefore, we expect that NeoCoV sooner may appear in human-related infection.
Keywords: 6 proteins; MERS-CoV; NeoCoV; SARS-CoV; bioinformatics analysis; coronaviruses; evolutionary study.
© The Author(s) 2020.
Conflict of interest statement
Declaration of conflicting interests:The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Figures











Similar articles
-
Detection and full genome characterization of two beta CoV viruses related to Middle East respiratory syndrome from bats in Italy.Virol J. 2017 Dec 19;14(1):239. doi: 10.1186/s12985-017-0907-1. Virol J. 2017. PMID: 29258555 Free PMC article.
-
Discovery of Novel Bat Coronaviruses in South China That Use the Same Receptor as Middle East Respiratory Syndrome Coronavirus.J Virol. 2018 Jun 13;92(13):e00116-18. doi: 10.1128/JVI.00116-18. Print 2018 Jul 1. J Virol. 2018. PMID: 29669833 Free PMC article.
-
Recombination analysis on the receptor switching event of MERS-CoV and its close relatives: implications for the emergence of MERS-CoV.Virol J. 2024 Apr 10;21(1):84. doi: 10.1186/s12985-024-02358-2. Virol J. 2024. PMID: 38600521 Free PMC article.
-
Severe acute respiratory syndrome vs. the Middle East respiratory syndrome.Curr Opin Pulm Med. 2014 May;20(3):233-41. doi: 10.1097/MCP.0000000000000046. Curr Opin Pulm Med. 2014. PMID: 24626235 Review.
-
Searching for animal models and potential target species for emerging pathogens: Experience gained from Middle East respiratory syndrome (MERS) coronavirus.One Health. 2017 Mar 3;3:34-40. doi: 10.1016/j.onehlt.2017.03.001. eCollection 2017 Jun. One Health. 2017. PMID: 28616501 Free PMC article. Review.
Cited by
-
ACE2-using merbecoviruses: Further evidence of convergent evolution of ACE2 recognition by NeoCoV and other MERS-CoV related viruses.Cell Insight. 2024 Jan 30;3(1):100145. doi: 10.1016/j.cellin.2023.100145. eCollection 2024 Feb. Cell Insight. 2024. PMID: 38476250 Free PMC article. Review.
-
Potential Therapeutic Target and Vaccines for SARS-CoV-2.Pathogens. 2023 Jul 10;12(7):926. doi: 10.3390/pathogens12070926. Pathogens. 2023. PMID: 37513773 Free PMC article.
-
Detection of Nonsynonymous Single Variants in Human HLA-DRB1 Exon 2 Associated with Renal Transplant Rejection.Medicina (Kaunas). 2023 Jun 9;59(6):1116. doi: 10.3390/medicina59061116. Medicina (Kaunas). 2023. PMID: 37374320 Free PMC article.
-
Comparative highlights on MERS-CoV, SARS-CoV-1, SARS-CoV-2, and NEO-CoV.EXCLI J. 2022 Sep 29;21:1245-1272. doi: 10.17179/excli2022-5355. eCollection 2022. EXCLI J. 2022. PMID: 36483910 Free PMC article. Review.
-
Exploring whole proteome to contrive multi-epitope-based vaccine for NeoCoV: An immunoinformtics and in-silico approach.Front Immunol. 2022 Aug 3;13:956776. doi: 10.3389/fimmu.2022.956776. eCollection 2022. Front Immunol. 2022. PMID: 35990651 Free PMC article.
References
-
- The International Committee for Taxonomy of Viruses (ICTV). http://talk.ictvonline.org/files/ictv_documents/m/msl/4090.aspx. Accessed June 27, 2014.
-
- Jain A, Mittal N, Sharma PC. Genome wide survey of microsatellites in ssDNA viruses infecting vertebrates. Gene. 2014;552:209-218. - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
